





Abstract
Recent advances in DNA sequencing technology have allowed the collection of high-dimensional data from human-associated microbial communities on an unprecedented scale. A major goal of these studies is the identification of important groups of microorganisms that vary according to physiological or disease states in the host, but the incidence of rare taxa and the large numbers of taxa observed make that goal difficult to obtain using traditional approaches. Fortunately, similar problems have been addressed by the machine learning community in other fields of study such as microarray analysis and text classification. In this review, we demonstrate that several existing supervised classifiers can be applied effectively to microbiota classification, both for selecting subsets of taxa that are highly discriminative of the type of community, and for building models that can accurately classify unlabeled data. To encourage the development of new approaches to supervised classification of microbiota, we discuss several structures inherent in microbial community data that may be available for exploitation in novel approaches, and we include as supplemental information several benchmark classification tasks for use by the community.
Introduction
Supervised classification is a machine learning approach for developing predictive models from training data. Each training data point consists of a set of input features (in this review, the relative abundance of taxa) and a qualitative dependent variable giving the correct classification of that data point. In microbiota analysis, such classifications might someday include disease states, therapeutic results, or forensic identification. The goal of supervised classification is to derive some function from the training data that can be used to assign the correct class or category labels to novel inputs (e.g. new samples), and to learn which features (here, taxa) discriminate between classes. Common applications of supervised learning include text classification, microarray analysis, and other bioinformatics analyses. For example, when microbiologists use the Ribosomal Database Project website to classify 16S rRNA gene sequences taxonomically, they are using a form of supervised classification (naïve Bayes) (Wang et al., 2007).
Machine learning methods are particularly useful for recognizing patterns in highly complex data sets such as human microbiota surveys. The human microbiota consists of about 100 trillion microbial cells, compared with our 10 trillion human cells, and these microbial symbionts contribute many traits to human biology that we would otherwise lack. For example, gastrointestinal microorganisms are involved in xenobiotic metabolism (Clayton et al., 2009), dietary polysaccharide degradation (Turnbaugh et al., 2009a,b; Hehemann et al., 2010), immune system development (Hooper et al., 2001), and a wide range of other functions. Compositional differences between microbial communities residing in various body sites are large, and comparable in size to the differences observed in microbial communities from disparate physical habitats (Ley et al., 2008). Understanding the diversity and distribution of the human microbiota, and especially the systematic changes that occur in different physiological and disease states, is predicted to have far-reaching effects on health and disease (Turnbaugh et al., 2007).
Each sample in a typical study of microbiota (using second-generation sequencing technology) contains hundreds or thousands of DNA sequences from an underlying community consisting of thousands of unique species-level operational taxonomic units (OTUs) (for a discussion of assignment of OTU clusters, see Schloss & Handelsman, 2005). Ecological assessments of such surveys have generally been restricted to measuring taxon relative abundances, analyzing within- and between-sample diversity (α and β diversity, respectively), exploring β-diversity patterns using unsupervised learning techniques such as clustering and principal coordinates analysis (PCoA), and performing classical hypothesis testing. These approaches may be limited in their ability to classify unlabeled data or to extract salient features from highly complex and/or sparse data sets. Fortunately, many techniques in supervised learning are designed specifically for those purposes. For example, supervised learning has been used extensively with success in microarray analysis, a field with similar dimensionality issues, to identify small groups of genes that can be used to distinguish between different types of cancer cells (Lee et al., 2005). These techniques may hold promise for future applications demanding a similar solution to microbial community classification, including medical diagnosis and forensics identification.
We now review some of the analyses that are typical of the current literature on the characterization of human microbiota, and then provide motivation for the application of supervised learning in this field of study. After introducing the benchmark classification tasks that we use in this review, we explore several possible constraints inherent in microbial community data that might aid researchers in choosing which type of models to use. In many cases where appropriate models already exist, we demonstrate their effectiveness by applying some examples to the benchmarks; in other cases, we suggest directions for research into novel approaches.
Common data types and recent examples from human microbiota analyses
Supervised classification requires training data, where each training sample has values for a number of independent variables, or features, and an associated classification label. In this review, we demonstrate that the taxon relative abundance vectors from 16S rRNA gene sequence surveys can serve as useful input features for some classification problems. Many of the techniques that we discuss are also applicable to metagenomic surveys, where the input features would be the abundances of thousands of functional genes. Other measurements of microbial community configuration could serve as useful input features as well; Lozupone & Knight (2008) describe many of these measurements in detail in a prior review in this journal. Typical results generated by human-associated microbial community analyses can be seen in Li et al. (2008), Wen et al. (2008), Grice et al. (2009), and often include the following components.
Analysis of taxon relative abundances
A common data structure in community ecological analysis is the sample-by-taxon abundance matrix. In addition to serving as the input for OTU-based measures of α and β diversity (described below), these matrices can be mined for taxa whose relative abundances vary significantly with sample type or treatment. For example, one robust finding involving differences in taxon relative abundances has been the association of obesity with gut microbiota that have a lower relative abundance of bacteria from the phylum Bacteroidetes (Turnbaugh et al., 2009a). In this review, we use sample-by-taxon abundance matrices as training data in our benchmark classification tasks. A common feature of such matrices is their data sparseness: most taxa are confined to a relatively small fraction of samples (high endemicity). Other data types, discussed below, may also serve as useful inputs in future supervised classification tasks, but will not be analyzed directly in this review.
α-Diversity analysis
Measures of α diversity (or, within-sample diversity) have a long history in ecology (Magurran et al., 2004). α-Diversity scores have been shown to be differentiated for communities from several types of human body habitat. For instance, skin-surface bacterial communities have been found to be significantly more diverse in females than in males (Fierer et al., 2008) and at dry sites rather than sebaceous sites (Costello et al., 2009; Grice et al., 2009), and the gut microbiota of lean individuals have been found to be significantly more diverse than those of obese individuals (Turnbaugh et al., 2009a). These studies suggest that in some cases α-diversity scores will be useful input features for building supervised classifiers.
β-Diversity analysis and clustering
β-Diversity analysis attempts to measure the degree to which membership or structure is shared between communities. Many classical metrics can be used to estimate the distance between communities, although those based on phylogenetic relatedness perform optimally in 16S rRNA gene-based surveys (Martin et al., 2002; Lozupone & Knight, 2008). A nonphylogenetic distance metric such as the common Euclidean distance treats all organisms as though they were equally related to one another, and thus it can fail to capture the similarity between two communities containing closely related organisms. This problem becomes especially important in microbial community analyses where individual species are not commonly shared across environments, such as on the human body.
Once measures of β diversity have been calculated, the entire data set may be visualized using one of several ordination methods, such as nonmetric multidimensional scaling or principal coordinates analysis (PCoA). PCoA performs a rotation of the intersample distance matrix (after centering) to represent those distances as accurately as possible in a small number of dimensions. Nonmetric ordination has a similar goal, but seeks to represent only the rank order of intersample distances, rather than the actual distances as in PCoA. After ordination, a reduced-rank approximation of the intersample distances can be visualized in two or three dimensions for exploratory analysis and for identifying samples that cluster by habitat or environmental factors. There is no reason to use only the first two or three dimensions, but the higher dimensions will represent increasingly subtle trends in the distance matrix. Another popular unsupervised method is to create a hierarchical clustering of samples, and to visualize the resulting tree. All of these approaches have the purpose of using a small number of dimensions to represent, as closely as possible, the actual differences between samples.
Numerous recent microbiota analyses have used sample clustering based on phylogenetic β-diversity metrics (e.g. unifrac) to explore compositional similarities between communities. Correlations include, for example, diet and phylogeny in mammal guts (Ley et al., 2008), body habitat, individual, and time in healthy adults (Costello et al., 2009), and fingertip microbiota on touched surfaces (Fierer et al., 2010). The latter case is particularly notable because it suggests that supervised classification based on phylogenetic β diversity might prove useful in future work in the field of forensic identification.
Supervised learning as an alternative
The main purpose of supervised learning is to build a model from a set of categorized data points that can predict the correct category membership of unlabeled future data. The category labels can be any type of important metadata, such as the disease state of the host. The ability to classify unlabeled data is useful whenever alternative methods for obtaining data labels are difficult (as in the use of microbial communities from the human body in forensic identification; Fierer et al., 2010) or potentially fatal (as in the use of gene expression profiles to classify cancer types; Lee et al., 2005). In this review, we generally restrict our discussions to classification problems where the labels are discrete (qualitative), but much of the content is applicable to regression problems where the labels are continuous (quantitative).
This goal of building predictive models is very different from the traditional goal of fitting an explanatory model to one's data set; here, we are concerned less with how well the model fits our particular set of training data, but rather with how well it will generalize to novel input data. Hence, we have a problem of model selection: we do not want a model that is too simple or general, because it will fail to capture subtle, but important information about our independent variables (underfitting), but we also do not want a model that is too complex or specific, because it will incorporate idiosyncrasies that are specific only to our particular training data (overfitting). What we really want to optimize is the expected prediction error (EPE) of the model on future data. For an extensive introduction to model selection and supervised learning, see Hastie et al. (2009a).
When the labels for our data are easily obtained, as in the classification of microbiota by body habitat where the body habitat is known (Costello et al., 2009), we have no use for a predictive model. In these cases, supervised learning can still be useful for building descriptive models of the data, especially in data sets where the number of independent variables or the complexity of their interactions diminishes the usefulness of classical univariate hypothesis testing. Examples of this type of model can be seen in the various applications of supervised classification to microarray data (Lee et al., 2005), in which the goal is to identify a small, but highly predictive subset of the thousands of genes profiled in an experiment for further investigation. In microbial ecology, the analogous goal is to identify a subset of predictive taxa. Of course, in these descriptive models, accurate estimation of the EPE is still important; that is how we know that the association of the selected taxa with the class labels is not just lucky or spurious. This process of finding small, but predictive subsets of features, called feature selection, will be of increasing importance as the size and dimensionality of microbial community analyses continue to grow.
A common way to estimate the EPE of a particular model is to fit the model to a subset (e.g. 90%) of our data and then test its predictive accuracy on the other 10% of our data. This gives us an idea of how well the model would perform on future data sets were we to fit it to our entire current data set. To improve our estimate of the EPE we can repeat this process 10 times so that each data point is part of the held-out validation data once. This procedure, known as cross-validation, allows us to compare models that use very different inner machinery or different subsets of input features. Of course if we try many different models and select the one that gives us the lowest cross-validation error for our entire data set, it is likely that our reported EPE will be too optimistic. This is similar to the problem of making multiple comparisons in statistical inference; some models are bound to get ‘lucky’ on a particular data set. Hence, whenever possible, we want to hold out an entirely separate test set for estimating the EPE of our final model, after performing model selection. We do just that for the benchmarks used in this paper: we randomly choose a fraction of the data to act as the test set; we use cross-validation within the remaining training set to perform model selection; and we report the prediction error of the final model when applied to the test set.
Even if we have established how to select the best parameters or degree of complexity for a particular kind of model, we are still faced with the problem of choosing what general class of models is most appropriate for a particular data set. The crux of choosing the right models for microbiota classification is to combine our knowledge of the most salient constraints (e.g. data sparseness) inherent in the data with our understanding of the strengths and weaknesses of various approaches to supervised classification. If we understand what structures are inherent in our data, we can then choose models that take advantage of those structures. For example, in the classification of microbiota, we may desire methods that can model nonlinear effects and complex interactions between organisms, or, due to the highly diverse nature of many microbial communities on the human body (Costello et al., 2009), we might want models designed specifically to perform aggressive feature selection when faced with high-dimensional data. Specialized generative models, discussed later in this review, can be designed to incorporate prior knowledge about the data as well as the level of certainty about that prior knowledge. Instead of learning to predict class labels based on input features, a generative model learns to predict the input features themselves. In other words, a generative model learns what the data ‘looks like’, regardless of the class labels. One potential benefit of generative models such as topic models (Blei et al., 2003) and deep layered belief nets (Hinton et al., 2006) is that they can extract useful information even when the data are unlabeled. We expect the ability to use data from related experiments to help build classifiers for one's own labeled data to be important as the number of publicly available microbial community data sets continues to grow.
So far there has been almost no application of machine learning classification techniques to microbial community data, according to an extensive literature search. One exception is an analysis of soil and sediment samples by Yang et al. (2006), in which the authors classified the samples according to environment type using support vector machines (SVMs) and k-nearest neighbors (KNN). Their data generally classified well, with an EPE of 0.04 for a set of Idaho soil samples, and an EPE of 0.14 for Chesapeake Bay samples, although they characterized communities using amplicon-length heterogeneity profiles rather than 16S rRNA gene-based taxon abundances or α/β-diversity measures. In contrast, supervised learning has been used extensively in other classification domains with high-dimensional data, such as macroscopic ecology (e.g. Cutler et al., 2007), microarray analysis (see above references), and text classification.
Benchmarks
While we do not intend this paper to be a comprehensive review of classification techniques, we do want to demonstrate that supervised classifiers can be effective and useful in microbiota analyses. To this aim, we used five benchmark classification tasks of varying size and difficulty involving actual human microbial communities. These data sets are included as supplemental information for the comparative evaluation of future approaches to supervised learning in this field. They are taken from two recent studies of human-associated microbial communities (Costello et al., 2009; Fierer et al., 2010). Both data sets were 16S rRNA gene surveys sequencing the V2 region with 454 pyrosequencing. After denoising each data set with the PyroNoise algorithm (Quince et al., 2009), we used the default settings in the qiime software package (Caporaso et al., 2010) to pick OTU clusters with uclust (Edgar et al., 2010) at a sequence similarity threshold of 97%. The choice of similarity threshold can have a significant effect on the quality of OTU abundances as predictive features, as we discuss later. In order to control for variable sequencing effort between samples, we performed a single rarefaction at the depth of the shallowest sample. There are several other preprocessing steps that require parameterization; we used the default settings in qiime, but a thorough benchmarking of the effects of various preprocessing choices on downstream analysis would be useful as a separate investigation.
α- and β-diversity analyses of the data are also likely to provide useful features for classification, although we constrain our discussion in this review to OTU abundances. For researchers wishing to perform a de novo analysis on these data sets, both have been made publicly available by the authors of the original studies. We now give details about the origin and purpose of each benchmark; Table 1 gives a summary of their sample sizes and dimensionality.
Summary of benchmark data sets used in this paper; singleton OTUs were removed
| Benchmark | Training samples | Test samples | No. OTUs | No. classes |
| Costello et al. Body Habitats (CBH) | 415 | 207 | 2741 | 6 |
| Costello et al. Skin Sites (CSS) | 268 | 133 | 2227 | 12 |
| Costello et al. Subject (CS) | 96 | 48 | 1592 | 7 |
| Fierer et al. Subject (FS) | 68 | 33 | 565 | 3 |
| Fierer et al. Subject × Hand (FSH) | 68 | 33 | 565 | 6 |
| Benchmark | Training samples | Test samples | No. OTUs | No. classes |
| Costello et al. Body Habitats (CBH) | 415 | 207 | 2741 | 6 |
| Costello et al. Skin Sites (CSS) | 268 | 133 | 2227 | 12 |
| Costello et al. Subject (CS) | 96 | 48 | 1592 | 7 |
| Fierer et al. Subject (FS) | 68 | 33 | 565 | 3 |
| Fierer et al. Subject × Hand (FSH) | 68 | 33 | 565 | 6 |
Summary of benchmark data sets used in this paper; singleton OTUs were removed
| Benchmark | Training samples | Test samples | No. OTUs | No. classes |
| Costello et al. Body Habitats (CBH) | 415 | 207 | 2741 | 6 |
| Costello et al. Skin Sites (CSS) | 268 | 133 | 2227 | 12 |
| Costello et al. Subject (CS) | 96 | 48 | 1592 | 7 |
| Fierer et al. Subject (FS) | 68 | 33 | 565 | 3 |
| Fierer et al. Subject × Hand (FSH) | 68 | 33 | 565 | 6 |
| Benchmark | Training samples | Test samples | No. OTUs | No. classes |
| Costello et al. Body Habitats (CBH) | 415 | 207 | 2741 | 6 |
| Costello et al. Skin Sites (CSS) | 268 | 133 | 2227 | 12 |
| Costello et al. Subject (CS) | 96 | 48 | 1592 | 7 |
| Fierer et al. Subject (FS) | 68 | 33 | 565 | 3 |
| Fierer et al. Subject × Hand (FSH) | 68 | 33 | 565 | 6 |
Benchmark 1: Costello et al. Body Habitats (CBH)
As noted above, microbial community composition tends to be highly differentiated between body habitats. The Costello et al. data included sample communities from six major categories of habitat: External Auditory Canal (EAC), Gut, Hair, Nostril, Oral cavity, and Skin. This benchmark is an example of a relatively easy classification task due to the generally pronounced differences between the communities, although some of the categories, such as Hair, are relatively under-represented. The benchmark excludes samples from communities that were transplanted from another subject or body site. We are subject here to the choice by the original authors to separate Hair and Nostril from Skin, when the three categories seem to be easily confused by the classifiers that we review. Of course in practice, these data would not normally require the use of a predictive model for classification, because the site of sampling would most likely be known. A more useful application for machine learning in this type of task is to perform feature selection to identify OTUs that are highly discriminative of the type of sampling site.
Benchmark 2: Costello et al. Skin Sites (CSS)
This benchmark is a subset of the full Costello et al. data, containing only those nontransplant samples taken from skin sites. The class labels are the specific type of skin site, and contain 12 unique classes (e.g. volar forearm, plantar foot, forehead, palm, etc.). The compositional differences between these categories are generally much more subtle than in the CBH benchmark; hence, the classification task is more difficult. As with the CBH benchmark, predictive models are not likely to be necessary for this particular classification task; the benchmark is instead intended to serve as a test bed for developing feature selection techniques as well as predictive techniques for use in other data sets where the category labels are more expensive to obtain.
Benchmark 3: Costello et al. Subject (CS)
This benchmark contains only a set of samples taken from the arms, hands, and fingers, excluding any ‘transplant’ samples. The class labels are the (anonymized) identities of seven of the nine subjects in the study. We omitted two of the subjects, ‘M5’ and ‘M6’, because they had very few samples. This benchmark is moderately challenging due the fact that samples come from heterogeneous time points (84 from June of 2008; 28 from September of that year). Costello et al. observed significant variation in individuals over time, and indeed although several classifiers achieved perfect expected accuracy when trained and tested only within the June samples, the lowest error achieved in our mixed test set was 0.062. In this case, predictive models (as opposed to descriptive models) may be more directly meaningful than in benchmarks CBH and CSS above: the ability to classify individuals by their microbiota could have the same applications in forensics as in the Fierer et al. data set discussed next.
Benchmark 4: Fierer et al. Subject (FS)
This benchmark contains all samples from the Fierer et al.‘keyboard’ data set (Fierer et al., 2010) for which at least 397 raw sequences were recovered (397 was chosen manually in order to include as many samples as possible). The class labels are the anonymized identities of the three experimental subjects, as with the CS benchmark above. This classification task is the easiest of all five benchmarks because of the clear distinctions between the individuals, because all of the samples come from approximately the same time point, and because of the large number of training samples available for each class.
Benchmark 5: Fierer et al. Subject × Hand (FSH)
This benchmark is a more challenging version of the previous one. The class labels are the concatenation of the experimental subject identities and the label of which hand (left vs. right) the sample came from on that individual. There were three subjects, and so there are six classes in this benchmark.
Test sets: For each of the five benchmarks, we have created 10 random splits of the data into training and test sets. A test set contains 1/3 of the data for a given benchmark, and the proportion of each class in the test set is approximately the same as its proportion in the overall data set. The indices of the test sets that we used in this paper are included with the benchmarks in the supplementary data.
Classifying classifiers
Several attempts have been made to review and organize published approaches to feature selection in high-dimensional classification problems, in some cases specifically with respect to microarray analysis, including, but not limited to (Forman, 2003; Guyon & Elisseeff, 2003; Man et al., 2004; Lee et al., 2005; Saeys et al., 2007). Lal et al. (2006) provide an excellent paradigmatic framework for categorizing and discussing the available techniques. The following section mentions a few issues in the design and application of classification methods relevant to microbial ecology; for a thorough taxonomy of classifiers, we refer the reader to the above articles.
Multiclass vs. binary
An important feature of a classifier is whether it can easily support multicategory (multiclass) classification. Some models, such as the original SVM, inherently support only binary decision problems. Other methods, such as KNN, multinomial logistic regression, and discriminant analysis allow for direct inference of multiclass decision boundaries. Binary classifiers can still be made to perform multiclass classification by collecting votes from one-vs.-one (pairwise) or one-vs.-all classifiers, but the lack of multiclass support becomes problematic when the number of classes is high, or when the data set is large.
Approaches to feature selection
As discussed earlier, the goal of feature selection is to find the combination of the model parameters and the feature subset that provides the lowest expected error on novel input data. We consider feature selection to be of utmost importance in the realm of microbiota classification due to the generally large number of features (i.e. constituent species-level taxa): in addition to improving predictive accuracy, reducing the number of features we use will help us to produce more interpretable models. Approaches to feature selection are typically divided into three categories: filter methods, wrapper methods, and embedded methods.
As the simplest form of feature selection, filter methods are completely agnostic to the choice of learning algorithm being used; that is, they treat the classifier as a black box. Filter methods use a two-step process. We first perform a univariate test (e.g. t-test) or multivariate test (e.g. a linear classifier built with each unique pair of features) to estimate the relevance of each feature, and select (1) all features whose scores exceed a predetermined threshold or (2) the best n features for inclusion in the model; then run a classifier on the reduced feature set. The choice of n can be determined using a validation data set or cross-validation on the training set.
Although filter methods may seem inelegant from a theoretical viewpoint due to their inherent lack of optimality, they are used extensively in the literature. They have several benefits, including their low computational complexity, their ease of implementation, and their potential, in the case of multivariate filters, to identify important interactions between features. The fact that the filter has no knowledge about the classifier is advantageous in that it provides modularity, but it can also be disadvantageous, as there is no guarantee that the filter and the classifier will have the same optimal feature subsets. For example, a linear filter (e.g. correlation-based) is unlikely to choose an optimal feature subset for a nonlinear classifier such as an SVM or a random forest (RF).
Wrapper methods are usually the most computationally intensive and perhaps the least elegant of the feature selection methods. A wrapper method, like a filter method, treats the classifier as a black box, but instead of using a simple univariate or multivariate test to determine which features are important, a wrapper uses the classifier itself to evaluate subsets of features. This leads to a computationally intensive search: an ideal wrapper would retrain the classifier for all feature subsets, and choose the one with the lowest validation error. Were this search tractable, wrappers would be superior to filters because they would be able to find the optimal combination of features and classifier parameters. The search is, however, not tractable for high-dimensional data sets; hence, the wrapper must use heuristics during the search to find the optimal feature subset. The use of a heuristic limits the wrapper's ability to interact with the classifier for two reasons: the inherent lack of optimality of the search heuristic, and the compounded lack of optimality in cases where the wrapper's optimal feature set differs from that of the classifier. We do not consider wrappers further in this review, because in many cases the main benefit of using wrappers instead of filters, namely that the wrapper can interact with the underlying classifier, is shared by embedded methods (discussed next), and the additional computational cost incurred by wrappers therefore makes such methods unattractive.
Research on embedded feature selection techniques has been plentiful in recent years, for example in Tibshirani (2002), Zou & Hastie (2005), and Lal et al. (2006). Embedded approaches to feature selection perform an integrated search over the joint space of model parameters and feature subsets so that feature selection becomes an integral part of the learning process. Embedded feature selection has the advantage over filters that it has the opportunity to search for the globally optimal parameter– feature combination. This is because feature selection can be performed with knowledge of the parameter selection process, whereas filter and wrapper methods treat the classifier as a ‘black box’. As discussed above, performing the search over the whole joint parameter–feature space is generally intractable, but embedded methods can use knowledge of the classifier structure to inform the search process, while in the other methods the classifier must be built from scratch for every feature set.
The classifiers discussed in this paper include several that perform embedded feature selection, several that use filter methods, and several that perform no explicit feature selection at all, but are nonetheless effective in high-dimensional data. The rest of this review is organized by several characteristics of microbial communities that we believe are important to consider when choosing between classification techniques. In cases where applicable techniques exist, we review several published examples; in other cases, we suggest directions for future work.
Review of selected classifiers
Microbial community data tend to be sparse; Fig. 1 shows a histogram of the frequency at which OTUs were observed in a given portion of samples in the Costello et al. data set. The full dataset consists of 816 samples and yields 14 254 OTUs when sequences are clustered with uclust at 97% similarity. The histogram in the figure excludes singletons, of which there were 10 471. Only 131 (0.9%) of the 14 254 observed OTUs were present in >10% of the samples; 97.7% of the species abundance matrix entries were zeros. Such high levels of sparsity are common in 16S rRNA gene microbial surveys, although the number of unique OTUs observed depends on several factors. The original sequencing data consists of millions of (generally) unique DNA sequences. In common practice, these sequences are binned into clusters at a predetermined similarity threshold. However, as shown in Quince et al. (2009), second-generation sequencing methods can be quite error-prone, and aggressive denoising is required to avoid having a falsely high number of so-called ‘distinct’ taxonomic groups, and so the OTUs in the Costello data shown in Fig. 1 were obtained after denoising with the PyroNoise algorithm. The choice of bin size (similarity threshold) for the step of picking OTUs has a notable effect on the discriminative value of the resultant OTUs, as will be discussed later in this paper.
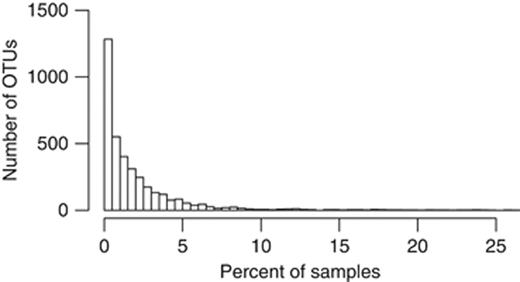
Histogram showing the number of OTUs that are present in a given percentage of samples for the full Costello et al. data set. Data were denoised using the PyroNoise algorithm, and OTUs were then picked at 97% similarity with the uclust software package. Of the 14 254 OTUs, 10 471 singletons were excluded before producing this histogram. This exemplifies the extreme sparsity typical of microbial community data sets.
The inherent sparseness of the abundance matrix is the fundamental challenge in building classifiers for microbiota; as discussed earlier, we know that systematic differences exist between many types of communities (such as between the gut communities of different human subjects), but identifying which OTUs will produce both good discrimination within the training data and good generalization to future test data remains challenging when so few OTUs are actually shared across communities. Although the following models have no explicit mechanism for incorporation of other kinds of prior knowledge, they are expected to perform well in high-dimensional classification problems such as those of concern, based on their published performance in other tasks with similar sparsity and dimensionality.
Random forests (RFs)
Although the RF classifier is not explicitly designed for performing feature selection or dimensionality reduction, it is one of the top performers in microarray analysis (Lee et al., 2005) as well as in many other domains with high-dimensional data (Breiman et al., 2001). RFs are an extension of bagging, or bootstrap aggregating, in which the final predictions of the model are based on an ensemble of weak predictors trained on bootstrapped samples of the data. An RF consists of many such classifiers, each of which is a decision tree. At each level of the decision tree, several randomly weighted linear combinations of small randomly selected subsets of features are evaluated by their ability to discriminate between categories, and the best subset is chosen to perform the split at that node. Other methods may outperform RFs in the presence of large numbers of irrelevant features (Gashler et al., 2008), but the strong performance of RFs in microarray analysis indicates that they should be effective for classifying at least moderately sized microbial communities. One drawback to RF is that it does not explicitly perform feature selection. It does provide a natural ranking of the relative importance of features (Breiman et al., 2001), but because most features are given at least some non-zero importance score we cannot easily identify the smallest number of features required to maintain a given level of accuracy.
Nearest shrunken centroids (NSCs)
The NSC classifier (Tibshirani et al., 2002) performed well on microarray data in an extensive comparative review (Lee et al., 2005). It is also fast, with algorithmic complexity scaling linearly in the number of features. NSC begins with the simplifying assumptions that an OTU's relative abundance is approximately normally distributed within each class, and that its abundance is independent of the abundance of other OTUs. Of course in general we might want to model the covariance of OTUs, but it may do more harm than good when we have many more OTUs than data points; in such cases, there are likely to be spurious correlations due to chance. In this simple model, we find, for each OTU, the mean within each class and the pooled within-class variance. This would give us an estimate of the location and spread of the centroid of all OTUs in each class. To classify a new sample, we would then calculate the log likelihood in each class of that sample's OTU abundance vector given the class centroids and the normality assumption, and then choose the class with the highest log likelihood.
Without any modification, this model is simply a linear discriminant analysis that assumes no covariation between OTUs (i.e. diagonal covariance). To instead perform feature selection and effectively denoise the centroid for each class, we first find the z-scores of the OTUs in each class centroid relative to the overall centroid. We then shrink all of these z-scores by a fixed amount λ, causing any with an absolute value of less than λ to become zero (i.e. we apply soft thresholding). The value of λ can be chosen using validation data or cross-validation within the training data. This gives us new shrunken z-scores for each OTU in each class. We map these back onto the overall centroid to get a shrunken centroid for each class, and then use these in place of the full centroids to classify new points as described above. The soft thresholding of the z-scores has the effect of zeroing out the least distinctive OTUs in each class. Hastie et al. (2009a) note that this procedure is basically applying a lasso-style penalty (see The elastic net) to the class z-scores.
The elastic net
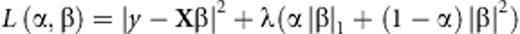
The ENET multinomial classifier has been shown to perform well on microarray data (Zou & Hastie, 2005). The fact that the ENET is capable of retaining groups of correlated input variables augurs well for its application to the classification of microbial communities, because in general we expect that some organisms have correlated patterns of abundance across communities. In the Costello et al. benchmark data set, for example, each OTU is on average highly correlated or anti-correlated (Pearson's coefficient of >0.5 or <−0.5) with 21.9 other OTUs (0.6%), and with 17.6 other OTUs (1.3%) in the Fierer et al. benchmark data set.
Support vector machines (SVMs)
SVMs also tend to be excellent all-around classifiers. The basic model is described in Cortes & Vapnik (1995). Traditional SVMs are restricted to binary classification tasks, although they are commonly applied to multiclass tasks by breaking the task into separate binary one-vs.-one or one-vs.-all tasks, and then allowing each model to vote for the final classification. SVMs have been effective in microarray classification tasks (Statnikov et al., 2005). The general approach taken by SVMs is to embed the n data points in an n-1 dimensional space in which the classes are linearly separable, and then to identify the hyperplane (known as the maximum-margin hyperplane), that maximizes the gap between the classes. This has the effect of minimizing the generalization error on unseen data. Choosing the right spatial embedding can allow an otherwise nonlinear class boundary to become linear, but in the case where the data are still not linearly separable, the SVM finds the maximum soft margin, where the objective function is penalized by some chosen cost function based on the distance of misclassified samples from the decision boundary. An SVM is so called because the separating hyperplane is supported (defined) by the vectors (data points) nearest the margin.
Although SVMs can perform poorly when given large numbers of irrelevant features, several approaches to feature selection combined with SVMs have proven successful in other high-dimensional classification problems, and hence these approaches may be useful ways to apply SVMs to microbial community data. In some studies, filter methods such as the ratio of the between-class sum-of-squares to the within-class sum-of-squares (BSS/WSS) have been effective for classifying microarray data or text when combined with SVMs (Lee et al., 2004; Lee et al., 2005). Other approaches use embedded feature selection, such as the zero-norm SVM, or R2W2 feature selection (Weston et al., 2001). More validation is needed for the zero-norm SVM, but it has been shown to perform well on one yeast classification experiment (Weston et al., 2003). R2W2 is simple, performs well on microarray test data, and tends to use a small number of features relative to other feature selection methods, although it does not support native multicategory classification. In this review, we use traditional SVMs both without filtering and with the three-filter methods discussed next.
Filter methods
The purpose of a filter is to identify features that are generally predictive of the response variable, or to remove features that are noisy or uninformative. Forman (2003) evaluates many common filters including the between-class χ2 test, information gain (decrease in entropy when the feature is removed), various standard classification performance measures such as precision, recall, and the F-measure, and the accuracy of a univariate classifier, among others. He also proposes a novel filter, called the bi-normal separation (BNS), which treats the univariate true-positive rate and the false-positive rate (tpr, fpr, based on document presence/absence in text classification) as though they were cumulative probabilities from the standard normal cumulative distribution function, and he uses the difference between their respective z-scores, F−1 (tpr)−F−1 (fpr), as a measure of that variable's relevance to the classification task. This approach is noteworthy because it outperformed all other filter methods in almost every performance measure that Forman reviewed, across several hundred test data sets. According to him, the BNS is effective because of the type of decision boundary it creates in the positive/negative document space of low-frequency words. More specifically, when compared with other methods, it tends to be just aggressive enough in avoiding rare words with mildly predictive tpr-to-fpr ratios. The fact that a single filtering method outperformed the others on an extensive and diverse suite of hundreds of experiments implies that a similar review in the domain of microbiota analysis may be illuminating.
The BNS filter will likely require some adaptation before it can be used on microbial community data, due to its reliance on the presence/absence of the feature in a given sample. For example, it is known in some cases that frequently occurring (i.e. nonsparse) microorganisms are associated with different clinical conditions (Turnbaugh et al., 2009a). Thus, the fact that the BNS uses presence/absence for determining true and false positive rates could cause common, but predictive OTUs to be ignored. It may be applicable in Forman's original formulation for extremely sparse datasets (high β and α diversity). The approach we followed in this review was to build univariate multiclass classifiers for all features, and to find the average true positive and false positive rates for each feature. These rates were then used to score features using BNS.
The second filter we consider is a type of backward feature elimination called recursive feature elimination, which was tailored to the SVM (SVM-RFE) by (Guyon et al., 2002). In SVM-RFE, we train a classifier using the full set of features (OTUs), remove the feature with the least influence on the current margin, and repeat with the reduced feature set until all features have been removed. The features are then ranked by importance in reverse order of removal.
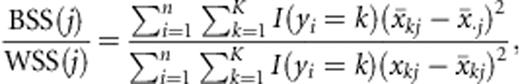
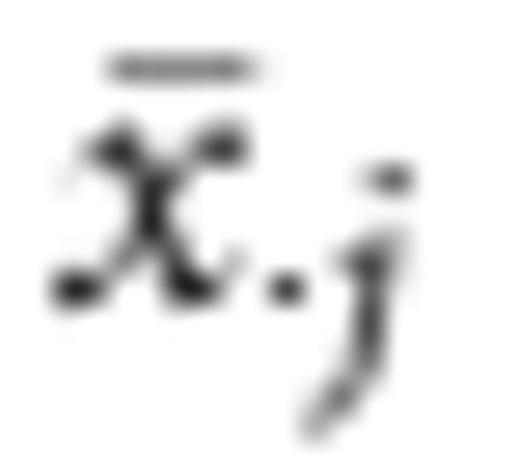 is the average value of the feature across all classes. The experiments in Lee et al. (2005) demonstrate that the BSS/WSS generally performs well, and the results that they obtain using the SVM with a radial basis kernel are comparable to those observed on the same data sets using embedded (nonfiltered) SVM approaches such as in (Weston et al., 2001).
is the average value of the feature across all classes. The experiments in Lee et al. (2005) demonstrate that the BSS/WSS generally performs well, and the results that they obtain using the SVM with a radial basis kernel are comparable to those observed on the same data sets using embedded (nonfiltered) SVM approaches such as in (Weston et al., 2001).Performance of selected classifiers on human microbiota
Table 2 contains the results of the unfiltered RF, NSC, ENET, and SVM classifiers on all of the benchmark data sets. For the four classifiers, we used publicly available implementations in the statistical software package r. Also included is the multinomial naïve Bayes (MNB) classifier, which is discussed later in the context of generative models. For each benchmark we report the number of features used by the models; in the case of RF we show the number of features with a non-zero importance score.
Performance of various classifiers on the benchmark data sets
| Method | Mean rank | Mean increase in error | Average test error (average number of OTUs) | ||||
| Costello Body Habitats | Costello Skin Sites | Costello Subject | Fierer Subject | Fierer Subject × Hand | |||
| RF | 1.7 | 0.01 | 0.09 (2484) | 0.34(2152) | 0.11 (1522) | 0.00(475) | 0.28 (507) |
| MNB | 2.3 | 0.05 | 0.08(2741) | 0.42 (2227) | 0.23 (1592) | 0.04 (554) | 0.23(554) |
| NSC | 2.4 | 0.04 | 0.09 (1842) | 0.42 (2006) | 0.20 (1391) | 0.01 (320) | 0.25 (326) |
| ENET | 3.6 | 0.06 | 0.11 (385) | 0.43 (700) | 0.13 (566) | 0.05 (59) | 0.33 (137) |
| SVM | 5.0 | 0.25 | 0.19 (2741) | 0.55 (2227) | 0.54 (1592) | 0.17 (554) | 0.54 (554) |
| Method | Mean rank | Mean increase in error | Average test error (average number of OTUs) | ||||
| Costello Body Habitats | Costello Skin Sites | Costello Subject | Fierer Subject | Fierer Subject × Hand | |||
| RF | 1.7 | 0.01 | 0.09 (2484) | 0.34(2152) | 0.11 (1522) | 0.00(475) | 0.28 (507) |
| MNB | 2.3 | 0.05 | 0.08(2741) | 0.42 (2227) | 0.23 (1592) | 0.04 (554) | 0.23(554) |
| NSC | 2.4 | 0.04 | 0.09 (1842) | 0.42 (2006) | 0.20 (1391) | 0.01 (320) | 0.25 (326) |
| ENET | 3.6 | 0.06 | 0.11 (385) | 0.43 (700) | 0.13 (566) | 0.05 (59) | 0.33 (137) |
| SVM | 5.0 | 0.25 | 0.19 (2741) | 0.55 (2227) | 0.54 (1592) | 0.17 (554) | 0.54 (554) |
For each classifier, for each benchmark, we show the mean test error over 10 repeated train/test iterations (standard errors (SEs) not shown), and the average number of features used in the final models produced over the 10 train/test iterations. Each train/test iteration consists of training the model on a randomly selected training set (training set sizes shown in Table 1), and then recording that model's error in predicting the labels for the unseen test set. The ‘mean rank’ column gives the average rank of that classifier across all benchmarks (lower is better); the rank of a classifier on a single benchmark is the standard fractional ranking. Fractional ranks were determined by considering models as tied when the better model's performance was within 1 SE of the worse model's performance. The ‘mean increase in error’ column gives the average difference between that model's test set error and the best model's test set error for a given benchmark (lower is better). Results in bold are within 1 SE of the best result for that column.
Performance of various classifiers on the benchmark data sets
| Method | Mean rank | Mean increase in error | Average test error (average number of OTUs) | ||||
| Costello Body Habitats | Costello Skin Sites | Costello Subject | Fierer Subject | Fierer Subject × Hand | |||
| RF | 1.7 | 0.01 | 0.09 (2484) | 0.34(2152) | 0.11 (1522) | 0.00(475) | 0.28 (507) |
| MNB | 2.3 | 0.05 | 0.08(2741) | 0.42 (2227) | 0.23 (1592) | 0.04 (554) | 0.23(554) |
| NSC | 2.4 | 0.04 | 0.09 (1842) | 0.42 (2006) | 0.20 (1391) | 0.01 (320) | 0.25 (326) |
| ENET | 3.6 | 0.06 | 0.11 (385) | 0.43 (700) | 0.13 (566) | 0.05 (59) | 0.33 (137) |
| SVM | 5.0 | 0.25 | 0.19 (2741) | 0.55 (2227) | 0.54 (1592) | 0.17 (554) | 0.54 (554) |
| Method | Mean rank | Mean increase in error | Average test error (average number of OTUs) | ||||
| Costello Body Habitats | Costello Skin Sites | Costello Subject | Fierer Subject | Fierer Subject × Hand | |||
| RF | 1.7 | 0.01 | 0.09 (2484) | 0.34(2152) | 0.11 (1522) | 0.00(475) | 0.28 (507) |
| MNB | 2.3 | 0.05 | 0.08(2741) | 0.42 (2227) | 0.23 (1592) | 0.04 (554) | 0.23(554) |
| NSC | 2.4 | 0.04 | 0.09 (1842) | 0.42 (2006) | 0.20 (1391) | 0.01 (320) | 0.25 (326) |
| ENET | 3.6 | 0.06 | 0.11 (385) | 0.43 (700) | 0.13 (566) | 0.05 (59) | 0.33 (137) |
| SVM | 5.0 | 0.25 | 0.19 (2741) | 0.55 (2227) | 0.54 (1592) | 0.17 (554) | 0.54 (554) |
For each classifier, for each benchmark, we show the mean test error over 10 repeated train/test iterations (standard errors (SEs) not shown), and the average number of features used in the final models produced over the 10 train/test iterations. Each train/test iteration consists of training the model on a randomly selected training set (training set sizes shown in Table 1), and then recording that model's error in predicting the labels for the unseen test set. The ‘mean rank’ column gives the average rank of that classifier across all benchmarks (lower is better); the rank of a classifier on a single benchmark is the standard fractional ranking. Fractional ranks were determined by considering models as tied when the better model's performance was within 1 SE of the worse model's performance. The ‘mean increase in error’ column gives the average difference between that model's test set error and the best model's test set error for a given benchmark (lower is better). Results in bold are within 1 SE of the best result for that column.
Using the randomforest package (Liaw & Wiener, 2002) with default settings, RF achieves the best performance of all classifiers, with the highest rank (inclusive of ties) for every benchmark. To evaluate the NSC classifier, we used the pamr package (Hastie et al., 2009b) with default settings. We see in Table 2 that NSC has fair performance on most of the benchmarks, but is clearly outperformed by the RF classifier in terms of test error and by the ENET classifier in terms of dimensionality reduction (i.e. reducing the number of OTUs required by the model). Using the ENET package glmnet (Friedman et al., 2010), and searching over 10 possible values for α (0.01, 0.1, 0.2, …, 0.9, 1.0) with otherwise default settings, we found that the ENET had somewhat higher prediction error on average than RF. In most cases, however, it drastically reduced the number of features used for the classification, and we found that the OTU subsets selected by the ENET tended to be good features for the RF classifier. For example, the 367 and 27 OTUs selected by the ENET for the CBH and FS benchmarks, respectively, allowed the RF classifier to obtain at least as high accuracy as with the full set of OTUs. While we do not know in general if these classifiers tend to agree about which features are important, the RF, NSC, and ENET classifiers had reasonable overlap for the FS benchmark. Supporting Information Fig. S1 shows a Venn diagram of the feature selection agreement between these three classifiers and the SVM-RFE filter (discussed below).
Figure 2 shows a heatmap plot of the 27 OTUs selected by the ENET for the FS benchmark. Using these OTUs, the RF classifier had 99.4% test accuracy across all test sets. Thus, these OTUs can be interpreted as representing the unique microbial ‘fingerprint’ of each individual. In the heatmap we see interesting systematic difference between individuals. The OTUs chosen by the ENET are quite diverse; it seems that each individual has a unique representation of OTUs across many bacterial families. Some of these may be related to distinct types of nonkeyboard surfaces that are commonly touched by each subject. For example, one subject appears to have a consistent over-representation of Pasteurellaceae, commonly found on mucosal surfaces of humans and animals (Kuhnert & Christensen, 2008). Another has very high relative abundances of Streptophyta, a plant phylum. It is important to note that this subset of features is not likely to be optimal in size or choice of OTUs for minimizing EPE; finding such a subset is intractable for all except for very small data sets. What we can say is that this is a highly predictive subset, capable of achieving perfect or near perfect accuracy on our benchmark test set.
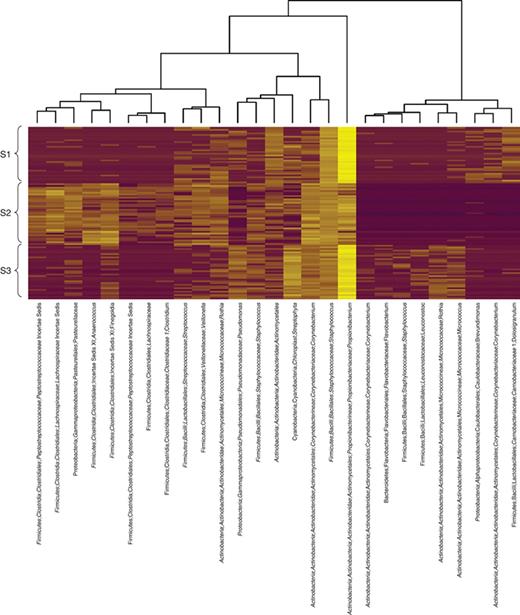
Heatmap of the log relative abundance of 27 OTUs forming the unique microbial fingerprint of each subject in the FSH benchmark. The OTUs were selected by the ENET classifier for assigning hand, fingertip, and keyboard microbial communities to the correct host. The ENET parameters (α, λ) were tuned using 10-fold cross-validation on the entire data set; using these parameters, the final model was then trained on the entire data set. OTU lineages were assigned by the Ribosomal Database Project classifier. Rows in the heatmap are standardized to zero mean and unit variance. Hierarchical clustering of columns was performed with Ward's method; rows were sorted by subject.
For the CBH benchmark, the OTUs selected by the ENET are representative of the previous findings related to human body microbiota reviewed above. Notably, the Oral cavity samples are distinguished by their relative abundance of Streptococcus, Pasteurellaceae, Prevotella, and Neisseria, and as expected, Bacteroides, Faecalibacterium, and Lachnospiraceae tend to be over-represented in samples from the gut. This result is a validation of the utility of supervised classifiers for selecting relevant features in a descriptive model. The heatmap of this OTUs subset is shown in Fig. S2.
For reviewing SVMs, we used the implementation in the ‘e1071’ package in r (Dimitriadou et al., 2010) with default settings (and the radial basis kernel). To optimize the cost and gamma parameters of the SVM, we performed a grid search over five values for each parameter and chose the combination that minimized cross-validation error within the given training set. We found that SVMs had consistently poor performance on the benchmarks when used without filtering. However, when combined with the SVM-RFE filter, SVM achieved similar performance to RF, with drastically smaller OTU subsets. The full results of the BSS/WSS, modified BNS, and SVM-RFE filters when combined with the RF and SVM classifiers are shown in Table 3. To obtain these filtered results, we first ranked the OTUs by each filter method, and then used the top n OTUs to build our final classifier, where we selected the n that minimized cross-validation error within the training set. This approach has led to very small feature sets with excellent accuracy on similar data sets (e.g. Guyon et al., 2002). The SVM-RFE and modified BNS results were all within 1 SE for both classifiers. Both classifiers performed better with a filter (Table 3) than without (Table 2), and a comprehensive study of filter methods applied to microbiota classification is recommended.
Performance of several classifiers on the FSH data set when combined with the BNS, BSS/WSS, and SVM-RFE filters
| Filter | Classifier | No. features | Test error |
| SVM-RFE | SVM | 40 | 0.27 |
| SVM-RFE | Random forests | 34 | 0.25 |
| BSS/WSS | SVM | 64 | 0.41 |
| BSS/WSS | Random forests | 52 | 0.28 |
| BNS | SVM | 70 | 0.29 |
| BNS | Random forests | 70 | 0.26 |
| Filter | Classifier | No. features | Test error |
| SVM-RFE | SVM | 40 | 0.27 |
| SVM-RFE | Random forests | 34 | 0.25 |
| BSS/WSS | SVM | 64 | 0.41 |
| BSS/WSS | Random forests | 52 | 0.28 |
| BNS | SVM | 70 | 0.29 |
| BNS | Random forests | 70 | 0.26 |
For each classifier–filter combination, the number of features was selected by leave-one-out cross-validation on the training set; this table reports the number of features and the test set error.
Performance of several classifiers on the FSH data set when combined with the BNS, BSS/WSS, and SVM-RFE filters
| Filter | Classifier | No. features | Test error |
| SVM-RFE | SVM | 40 | 0.27 |
| SVM-RFE | Random forests | 34 | 0.25 |
| BSS/WSS | SVM | 64 | 0.41 |
| BSS/WSS | Random forests | 52 | 0.28 |
| BNS | SVM | 70 | 0.29 |
| BNS | Random forests | 70 | 0.26 |
| Filter | Classifier | No. features | Test error |
| SVM-RFE | SVM | 40 | 0.27 |
| SVM-RFE | Random forests | 34 | 0.25 |
| BSS/WSS | SVM | 64 | 0.41 |
| BSS/WSS | Random forests | 52 | 0.28 |
| BNS | SVM | 70 | 0.29 |
| BNS | Random forests | 70 | 0.26 |
For each classifier–filter combination, the number of features was selected by leave-one-out cross-validation on the training set; this table reports the number of features and the test set error.
Mining phylogenetic relationships
It is possible to use a global alignment of the DNA sequences belonging to the different OTUs in a collection of microbial communities to place those OTUs in a phylogenetic tree. This tree has the potential to provide much more information about the similarity of communities than the raw counts of OTUs, as the tree allows us to measure the similarity of two communities by how closely related their constituent taxa are. In contrast, using only the raw relative abundance of OTUs to calculate intercommunity distance assumes that all OTUs are equally related to one another (i.e. related by a ‘star’ phylogeny).
Phylogenetic distance measures that use the structure of the tree have been shown to recover known clusters of microbial communities in data sets where nonphylogenetic distance measures fail (Lozupone & Knight, 2005). For example, Fig. 3 shows sample scores on the first two PCoA axes of the intersample distances in the CBH benchmark using phylogenetic (unifrac) and nonphylogenetic (Bray– Curtis) distance metrics. The points in each plot represent individual microbial communities, and the colors represent the body sites from which the samples were taken. The phylogenetic distance metric clearly shows much better clustering of the samples by body site than the nonphylogenetic distance metric. Phylogenetic analysis is almost certain to provide useful derived features for supervised learning in some cases, although how best to mine the phylogenetic relationships for useful features is an open question.
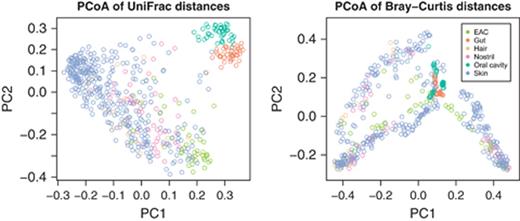
Left: the first two principal axes of PCoA of body habitat samples from the CBH benchmark based on unifrac (phylogenetic) distances; Right: the same analysis using Bray–Curtis (nonphylogenetic) distances. Note that the phylogenetic distance measure shows a clear separation of the Gut and Oral Cavity samples from the rest of the samples, while the nonphylogenetic distance measure places them approximately in the center of the distribution.
Phylogenetic depth of OTUs
As discussed earlier, the raw data produced in a 16S rRNA gene-based survey consists of millions of (generally) unique nucleotide sequences. In order to facilitate analysis, these sequences are commonly binned into clusters based on similarity at a predetermined similarity threshold. In this paper, we use the default settings of the qiime software package for picking OTUs (Caporaso et al., 2010). By default, qiime uses uclust for picking OTUs at 97% sequence similarity, but the choice of similarity threshold may provide a natural source of dimensionality reduction: as we lower the similarity threshold, the bins get larger, and we get fewer OTUs. Figure 4 shows the average test error for the RF classifier on the FSH benchmark for each of 10 random train/test splits as we varied the level of similarity within OTU clusters. Quite surprisingly, the expected performance of the classifier is about the same at all levels of similarity between 65% and 95%, with almost a 100-fold range in dimensionality. That is, for the FSH benchmark a model built using 14 very general OTUs is just as effective on average (although with a bit higher variance across training sets) as a model built using 1282 very specific OTUs. It is also interesting to note that for this classification problem, accuracy gets noticeably worse at very high levels of similarity such as 97% and 99%, suggesting that for some data sets, too much specificity makes it difficult for the RF classifier to capture broad trends at higher taxonomic levels.
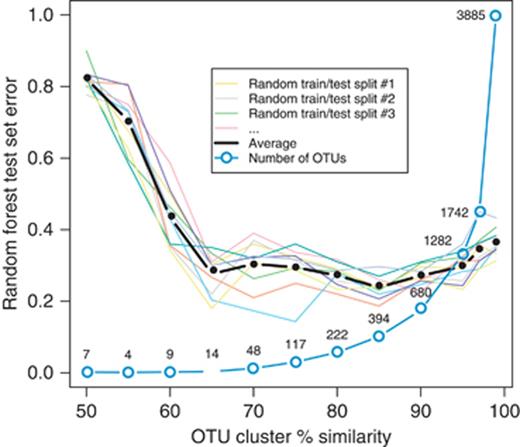
RF test set error as the percent similarity threshold for building OTU clusters is varied using the uclust software package. Colored lines show the results for 10 randomly chosen splits of the data into training and test sets; the thick black line shows the average of all 10. Also shown (in blue circles) is the number of OTUs chosen at each similarity level. Note that the classifier has approximately equivalent accuracy with 14 very general OTU clusters as it does with 1282 very specific OTUs clusters.
Clearly, the issue of feature selection or dimensionality reduction in microbiota analyses is in some cases intimately tied to the taxonomic specificity of our OTUs. However, there are certain known phyla for which subtle genetic differences even between different strains of a species can make the difference between pathogen and nonpathogen, such as in the case of Pseudomonas aeruginosa (Van Eldere, 2003); hence, it cannot be the case that we always want to reduce dimensionality by reducing taxonomic specificity. It may be that hybrid models using several levels of phylogenetic binning will outperform those constrained to any one bin size, and this is certainly an area that requires further research.
Metabolic functions as latent factors
OTUs that are relatively exchangeable with one another in terms of functional (metabolic) behavior may not be present in the same communities due to competitive exclusion (Horner-Devine et al., 2007). Therefore, it may not always make sense to do feature selection with OTUs, especially when we are dealing with highly specific OTU clusters. If indeed our classification categories are differentiated by the functional behavior of their communities rather than by the specific species-level taxa they comprise, then what we really want to learn is a set of functional equivalence classes, each containing a set of functionally redundant OTUs. We can then carry out inference in the reduced space of the latent functional profiles that are generating the observed community structures, rather than in the much more complex space of the OTUs that are performing those functions. Recent advances in design and inference of complex generative models such as deep belief nets and the many derivatives of topic models may allow us to recover these simple latent factors from the relatively complex communities that we observe.
We may consider the following as a simple generative model for microbial communities: each environment can be viewed as a weighted mixture of (i.e. multinomial distribution over) metabolic functions, where each function is performed by a weighted mixture of species. If all communities in a given data set draw from the same set of metabolic functions and the same set of species, this generative model is known as latent Dirichlet allocation (LDA), a popular model from the field of natural language processing introduced in Blei et al. (2003). LDA was originally used for automatically extracting conversation topics in the unsupervised semantic analysis of text. If each microbial community is treated as though it were a separate text document in a corpus of documents, the semantic topics in topic modeling are analogous to the metabolic functions or pathways that occur in the communities, and the vocabulary words correspond to OTUs. LDA is a purely generative model; it seeks only to model the distribution of the observed data, P(D), rather than learning to predict class labels based on the data, P(L|D). For the purposes of classification, we would of course need to incorporate some discriminative learning into the model. The simplest approach is to ‘piggy-back’ a generic classifier such as RF on top of LDA, using the distribution over latent functions in each community resulting from LDA inference as input features instead of, or in addition to, the raw OTU counts. This approach was used for text classification in Blei et al. (2003) and has the potential to work well when the differences between our classification categories are the most important determinant of the mixing proportions of latent functions.
A more direct and more powerful approach is to learn explicitly the joint distribution over category labels and data, P(L,D). This has the potential to combine the strengths of generative and discriminative learning. Several such supervised versions of LDA have been developed, such as multiconditional learning (MCL) (McCallum et al., 2006) and supervised LDA (SLDA) (Blei & McAuliffe, 2008). SLDA is the most general, being applicable to many types of response variable including categorical labels (classification) and real-valued labels (regression). MCL is restricted to classification tasks, but was shown by the authors above to perform well in a large variety of text classification problems.
To encourage the evaluation of these types of generative and hybrid (i.e. generative and discriminative) models in future research on microbiota analysis, we show evidence that the latent mixtures over OTUs recovered by classical LDA are indeed related to the category labels in the FSH benchmark. In Fig. 5, we show the test set error of an RF classifier using as features the per-community OTU mixtures learned with LDA, plotted against the log-likelihood, given the inferred topic model, of the entire OTU abundance matrix. Each data point was obtained by choosing random values for the LDA model's hyperparameters α, η, uniformly from the interval [0.1, 0.5], and then performing collapsed Gibbs sampling to infer a topic model with 25 topics using the lda package for r (Chang et al., 2010). An increase in quality of the fit obtained by the model, as measured by the corresponding log likelihood, is clearly a general indication of an increase in quality of the inferred features as predictors of the class labels (Pearson's correlation coefficient=−0.51, P-value=2 × 10−16).
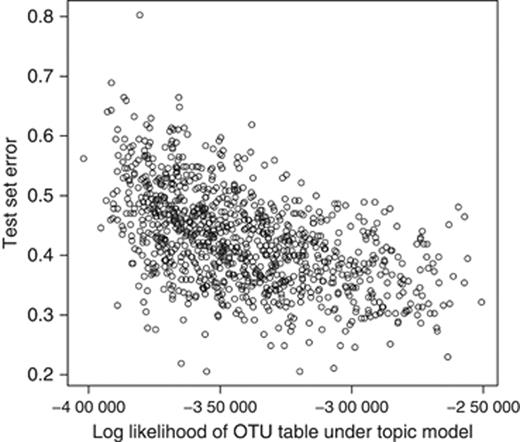
RF test set error plotted against the log likelihood of the data given a particular topic model. Each data point represents one topic model trained on the entire FSH benchmark data set using randomly chosen values of the topic model's hyperparameters. The latent ‘topics’ recovered by the topic model were then fed into the RF classifier as the only input features. While the error rates here are no better than those in Fig. 4 or Table 2, the correlation between the generative log likelihood and the discriminative ability of the derived latent features (topics) implies that topic models may be appropriate generative models for microbial communities; the better the topic model does at modeling the data, the more useful the inferred topics are for explaining differences between communities.
We also included a simple MNB classifier (McCallum & Nigam, 1998) in our comparison of classifiers, as shown in Table 2. MNB is the equivalent of a labeled topic model where each class has one topic (mixture of OTUs) that is shared by all of its samples, and where we learn the topics' mixture components conditioned on the class labels. Our implementation of MNB includes a small prior count for each OTU in each class to act as a smoothing constant, and we chose the value of this constant that minimized cross-validation error within the training set. Although this is a very simple model, it has performance competitive with RF. When compared with the RF, NSC, ENET, and SVM classifiers, MNB achieved the second best mean rank.
Data augmentation
Existing high-throughput experiments in microbial ecology typically have many fewer samples than observed species-level taxa, making it difficult to model complex interactions between the taxa. However, as the number of published experiments grows, there are an increasing number of data sets available that can potentially be used as unlabeled data to augment the labeled training data in a given experiment. Even relatively different data sets may still be useful for training generative and hybrid generative/discriminative models if they contain information about similar OTUs, or even about the ways in which OTUs interact.
For our five benchmarks, we found that adding noisy replicates of training data (Lee et al., 2000; Nair & Hinton, 2009) tends to be effective at increasing the predictive power of our models. For each of the benchmark data sets, we generated noisy replicates of the training data by adding a small amount of Gaussian noise (with mean zero and variance equal to the average within-sample variance) to the counts of OTUs present in each sample, thresholding the resulting counts at zero to avoid negative abundance values. We added three noisy replicates of the training set to itself, and fed the augmented train set to an RF classifier with 500 trees. In all cases, the EPE when using the augmented training set was as good as or better than that of the best unaugmented model, although the differences were on the order of a 1% or 2% decrease in error. Of course we encourage researchers interested in building supervised classifiers to collect as many samples as possible for a training set, but for cases where there is insufficient training data available, we suggest the exploration of augmented training data, both in the form of noisy training sample replicates and in the form of unlabeled samples from related microbial communities, as an important direction for future research. Such multisourced experimentation presents its own challenges; it would at least require uniform labeling of samples (metadata). Metadata standardization efforts such as those by the Genomic Standards Consortium (e.g. Field et al., 2008) will be essential for large-scale multisourced data augmentation.
Concluding remarks
Supervised learning can serve several purposes for researchers who wish to characterize differences between microbiota in different types of communities. In experiments where the true category membership of communities is well-known or is easily obtained, sparse classification techniques such as filter methods or the ENET can be used to identify specific taxa that are highly discriminative of the categories. The RF classifier may be useful in these cases as well; although it does not explicitly perform any dimensionality reduction, it produces a natural ranking of features by their importance in the model, and it tends to have lower EPE than the other models. In other classification tasks such as forensic identification or the early prediction of disease states, supervised classifiers could be used to learn a predictive model that generalizes well to unseen data. For example, as the cost of DNA sequencing continues to decline, it may become possible to perform gut microbiota surveys of all individuals in a diseased population in order to recommend personalized therapy (Turnbaugh et al., 2009b). In such cases, where class prediction is the ultimate goal, one should simply choose whatever model gives us the lowest EPE whether or not it performs explicit feature selection.
We presented five benchmark classification tasks containing data from bacterial 16S rRNA gene-based surveys of various human body habitats. The benchmarks contain classification tasks of varying difficulty, ranging from distinguishing individual humans by their hand microbiota, which can be done with perfect accuracy, to distinguishing different types of skin sites across individuals, on which task the best classifier we evaluated has 26% expected generalization error. We have made available the same benchmarks as a resource for those interested in pursuing novel techniques for microbiota classification.
All of the supervised classifiers that we reviewed have performed well in similar domains such as microarray analysis or text classification, but it is clear from their performance on our benchmarks that some perform better than others in microbiota classification. RFs was clearly the strongest performer, being tied for first place in all five of the benchmarks. MNB also tended to perform well, suggesting that generative models like SLDA may be worth exploring. SVMs had surprisingly poor performance without filtering, but they seemed to combine well with the BSS/WSS and SVM-RFE filters. The ENET classifier tended to have noticeably higher expected error than RFs, although it still proved useful for performing feature selection as a preprocessing step for other classifiers. For example, we included a heatmap of the 27 OTUs selected by the ENET classifier in the FS benchmark. These OTUs allow >99% test accuracy when trained with the RF classifier, and thus they represent the unique microbial ‘fingerprint’ of each individual.
Future research into approaches that leverage natural structures inherent in the microbial community data is strongly recommended. Examples include performing dimensionality reduction by reducing the phylogenetic specificity of taxonomic clusters, utilizing the naturally hierarchical structure of features provided by phylogenetic trees, using related data sets as unlabeled data to aid in the inference of generative models, and the exploration of generative or hybrid generative/discriminative techniques to recover latent features, such as metabolic functions, that drive the differences in observed taxa across communities. However, existing classifiers perform well for a range of tasks and will be widely useful in human microbiome projects, perhaps, especially, for identifying biomarkers for disease or other physiological conditions.
References
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Editor: Mecky Pohlschroder

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}