





Abstract
Members of the genus Pseudomonas inhabit a wide variety of environments, which is reflected in their versatile metabolic capacity and broad potential for adaptation to fluctuating environmental conditions. Here, we examine and compare the genomes of a range of Pseudomonas spp. encompassing plant, insect and human pathogens, and environmental saprophytes. In addition to a large number of allelic differences of common genes that confer regulatory and metabolic flexibility, genome analysis suggests that many other factors contribute to the diversity and adaptability of Pseudomonas spp. Horizontal gene transfer has impacted the capability of pathogenic Pseudomonas spp. in terms of disease severity (Pseudomonas aeruginosa) and specificity (Pseudomonas syringae). Genome rearrangements likely contribute to adaptation, and a considerable complement of unique genes undoubtedly contributes to strain- and species-specific activities by as yet unknown mechanisms. Because of the lack of conserved phenotypic differences, the classification of the genus has long been contentious. DNA hybridization and genome-based analyses show close relationships among members of P. aeruginosa, but that isolates within the Pseudomonas fluorescens and P. syringae species are less closely related and may constitute different species. Collectively, genome sequences of Pseudomonas spp. have provided insights into pathogenesis and the genetic basis for diversity and adaptation.
Introduction
Members of the genus Pseudomonas (sensu stricto) show remarkable metabolic and physiologic versatility, enabling colonization of diverse terrestrial and aquatic habitats (Palleroni et al., 1992), and are of great interest because of their importance in plant and human disease, and their growing potential in biotechnological applications (Fig. 1). Since the genus Pseudomonas was first described, the assignment of isolates to and within the genus has been contentious. Stanier et al. (1966) published a comprehensive appraisal of the taxonomy of Pseudomonas spp., largely determined by phenotypes and biochemical capabilities. The resolution of its intrageneric structure using DNA–DNA hybridization (DDH), analysis of rRNA and housekeeping gene sequences and multilocus sequencing (Palleroni et al., 1973; Moore et al., 1996; Maiden et al., 1998; Gardan et al., 1999; Anzai et al., 2000; Yamamoto et al., 2000; Goris et al., 2007) have aided taxonomic definition and species reorganization (Palleroni & Moore, 2004). Based largely on data from molecular studies, strains thought to belong to Pseudomonas sensu stricto (in the Gamma-subclass of Proteobacteria) have been separated from the genus and placed into the genera Burkholderia, Ralstonia and Comamonas (Betaproteobacteria) (Kersters et al., 1996). Whole-genome sequences provide an opportunity to further address the systematics of the genus.
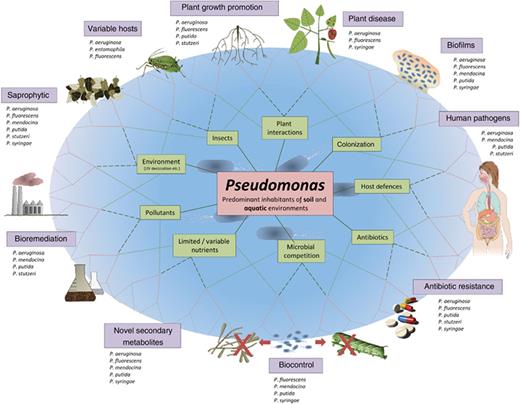
The functional and environmental range of Pseudomonas spp. The Pseudomonas common ancestor has encountered a wide range of abiotic and biotic environments that has led to the evolution of a multitude of traits and lifestyles with significant overlap among species.
Pseudomonas aeruginosa PAO1 was the 25th bacterial genome sequence to be completed (Stover et al., 2000). Genome sequences of strains of Pseudomonas syringae (Buell et al., 2003), Pseudomonas putida (Nelson et al., 2002), Pseudomonas fluorescens (Paulsen et al., 2005), Pseudomonas entomophila (Vodovar et al., 2006), Pseudomonas mendocina (unpublished data) and Pseudomonas stutzeri (Yan et al., 2008) followed, and have since been augmented by additional sequences of several strains of these bacterial species. The genome of the nitrogen-fixing bacterium Azotobacter vinelandii, classified in the Pseudomonadaceae, has also been sequenced (Setubal et al., 2009). In each case, genome sequencing was undertaken to identify the genetic features contributing to the particular lifestyle or phenotype of interest for the sequenced isolate. As of January 2011, there were 18 complete Pseudomonas genomes listed in NCBI's Entrez database, with another 72 listed as being draft assemblies or incomplete. Of these 90, 15 are P. aeruginosa isolates (strain PA14 is listed in both complete and draft project pages) and 38 are pathovars of P. syringae (strain B728a is listed in both complete and draft project pages). That close to 65% of sequences are derived from just two pathogenic members of the genus demonstrates the considerable importance of these Pseudomonas spp. in human and plant health.
Here, we review the genomes of Pseudomonas spp., first considering the genome structures of the bacteria. These are considered in the context of the lifestyle and properties of Pseudomonas spp. with an emphasis on diversity and adaptability. We consider the potential uses of genomics for studies of systematics and evolution. We shall not specifically review the genomics of secondary metabolite production. Instead, we direct readers to an excellent review by Gross & Loper (2009).
Genome sequence features
‘Core’ and ‘accessory’ genomes and the conserved gene set
The concept of a ‘core’ genome has been used to describe the conserved sequences in P. aeruginosa, with the remaining genes comprising the accessory genome, which has been defined as the set of genes missing from one or more strain (Mathee et al., 2008). Because the core group of genes is refined each time a new genome sequence is released, the core gene complement is defined as the conserved set of genes common to a particular group of organisms under consideration. This core set will reduce in size as additional genome sequences are determined, and it is not possible to know how many sequences will be required before a ‘plateau’ is reached where the true core gene complement can confidently be assigned. Ignoring the large inversions found in some P. aeruginosa strains, the regions of conserved genes could be mapped onto the PA14 genome as a template, revealing the location and size of strain-specific segments, many of which originate from horizontal gene transfer (Mathee et al., 2008).
Genomic islands (GIs), phage and plasmids – the accessory genome in Pseudomonas spp.
Mobile genetic elements (MGEs) such as phages, plasmids, transposons and GIs are frequently identified in microbial genomes, and can play significant roles in processes such as pathogenesis and antibiotic resistance (Arnold et al., 2003; Hacker et al., 2004; Jackson et al., 2011). In Pseudomonas spp., MGEs have been identified in all species in which they have been sought. While these often follow the established patterns (e.g. integration into tRNA genes), there is considerable variability in the identity, location and functional gene content, even within strains of a given species. Most species have at least one prophage-like element (some may be functional while others are degraded remnants), several coding sequences (CDSs) with similarity to transposase genes and plasmid-related sequences. The influence of MGEs on Pseudomonas genomes is considerable in some cases, but appears to be less prominent in P. entomophila (Vodovar et al., 2006) and P. stutzeri (Yan et al., 2008), although key nitrogen-fixation functions in the latter are encoded on a GI. The accessory genome will be described in more detail later in this review.
Repetitive extragenic palindromic (REP) elements
These DNA sequences vary in length between 21 and 65 bp and are found in extragenic regions of the genomes of some bacteria (Tobes & Pareja, 2006). Their role is not precisely defined, but because they can stabilize mRNA and are the target for DNA polymerase I, DNA gyrase, integration host factor and transposases and recombinases, they may play a significant role in genome evolution and adaptation (reviewed in Tobes & Pareja, 2006). REP sequences are discussed throughout this review.
Plant growth promotion and bioremediation
Many pseudomonads interact with plants and several species contribute to plant health by antagonizing plant-pathogenic microorganisms (biocontrol) and directly influencing plant disease resistance and growth (plant growth promotion) – both as plant endophytes (Ryan et al., 2008) and as rhizosphere colonizers (Haas & Defago, 2005). Major secondary metabolites produced by Pseudomonas strains have been detected and investigated because of their antimicrobial activity (Leisinger & Margraff, 1979) and include 2,4-diacetylphloroglucinol (Raaijmakers et al., 1997), pyoluteorin (Howell & Stipanovic, 1980), pyrrolnitrin (Howell & Stipanovic, 1979), hydrogen cyanide (Voisard et al., 1989), syringomycin (Sorensen et al., 1996), syringopeptin (Lavermicocca et al., 1997), viscosinamide (Nielsen et al., 1999), viscosin (Neu et al., 1990; de Bruijn et al., 2007), thioquinolobactin (Matthijs et al., 2007), phenazines (Thomashow & Weller, 1988) and yet to be identified compounds (Garbeva & de Boer, 2009). Pseudomonads are also able to produce insecticides, including P. fluorescens Pf-5 Fit (for P. fluorescens insecticidal toxin) against tobacco hornworm (Péchy-Tarr et al., 2008); P. entomophila with currently unknown toxin(s) against Drosophila (Vodovar et al., 2005); and P. syringae with an unknown action against aphids (Stavrinides et al., 2009).
Bioremediation uses microorganisms to degrade or detoxify hazardous environmental contaminants. The exceptional nutritional versatility of pseudomonads, coupled with the production of biosurfactants that can mobilize hydrocarbons and nonaqueous phase liquids into an aqueous phase (Desai & Banat, 1997), makes them excellent candidates for bioremediation. Pseudomonas aeruginosa is frequently isolated in petroleum-contaminated soils and groundwater (Ridgway et al., 1990). Pseudomonas putida has been extensively studied in environmental biotechnology because of its capabilities in the bioremediation of toxic organic wastes including aromatic hydrocarbon compounds (Loh & Cao, 2008). Other Pseudomonas species identified with bioremediation properties include P. mendocina (Whited & Gibson, 1991) and P. stutzeri (Grimberg et al., 1996).
Several studies have used screening technologies to identify genes important for soil, and plant root (rhizosphere) and leaf (phyllosphere) colonization (Rainey et al., 1999b; Boch et al., 2002; Gal et al., 2003; Rediers et al., 2003; Silby & Levy, 2004; Marco et al., 2005; Ramos-Gonzalez et al., 2005). However, most of these screens were limited in scope because many of the genetic screens were proof of principle and did not screen the genome to saturation, thus only providing limited genome coverage. A major driver for sequencing the genomes of these bacteria was to gain a deeper insight into bacterial function, both in terms of their niche colonization and their metabolic and physiological properties.
Pseudomonas putida KT2440 – a biodegradation bacterium
Pseudomonas putida are soil- and plant-associated bacteria of interest for their biodegradation properties. Like most other Pseudomonas spp., P. putida genomes are larger than 6 Mbp. Annotation of the P. putida genome sequences (Tables 1 and 2) showed a considerable number of CDSs of unknown function, which is typical for Pseudomonas genomes. A comparison of P. putida KT2440 with the P. aeruginosa PAO1 genome indicated that 85% of the CDSs were homologous (Nelson et al., 2002). Interestingly, 508 CDSs (9.4%) were identified as putative duplications, almost double that seen for P. aeruginosa. The largest family of duplications consists of 41 transposase genes, seven of which were novel to P. putida. These data suggest that gene duplication and transposable elements were of considerable importance in shaping the genome evolution of P. putida.
General features from completed Pseudomonas genomes
| Species/strain | Size | % G+C | Genes | % coding | tRNA | rRNA | Released | GenBank | Reference |
| P. aeruginosa | |||||||||
| PAO1 | 6 264 404 | 66.6 | 5671 | 89.8 | 63 | 13 | 13/09/2000 | AE004091.2 | Stover et al. (2000) |
| PA7 | 6 588 339 | 66.4 | 6396 | 90.1 | 65 | 12 | 05/07/2007 | CP000744.1 | Roy et al. (2010) |
| UCBPP-PA14 | 6 537 648 | 66.3 | 5994 | 89.8 | 63 | 13 | 06/10/2006 | CP000438.1 | Lee et al. (2006) |
| LESB58 | 6 601 757 | 66.3 | 6026 | 88.9 | 67 | 13 | 24/12/2008 | FM209186.1 | Winstanley et al. (2009) |
| C3719 | 6 222 097 | 66.5 | 5696 | 86.6 | 40 | 6 | 04/01/2006 | NZ_AAKV00000000 | Mathee et al. (2008) |
| PA2192 | 6 905 121 | 66.2 | 6203 | 85.5 | 44 | 2 | 04/01/2006 | NZ_AAKW00000000 | Mathee et al. (2008) |
| P. entomophila | |||||||||
| L48 | 5 888 780 | 64.2 | 5293 | 89.8 | 78 | 22 | 10/05/2006 | CT573326.1 | Vodovar et al. (2006) |
| P. fluorescens | |||||||||
| Pf0-1 | 6 438 405 | 60.6 | 5741 | 90.0 | 73 | 19 | 07/10/2005 | CP000094.2 | Silby et al. (2009) |
| Pf-5 | 7 074 893 | 63.3 | 6144 | 88.7 | 71 | 16 | 30/06/2005 | CP000076.1 | Paulsen et al. (2005) |
| SBW25 | 6 722 539 | 60.5 | 6009 | 88.3 | 66 | 16 | 09/01/2008 | AM181176.4 | Silby et al. (2009) |
| P. mendocina | |||||||||
| ymp | 5 072 807 | 64.7 | 4730 | 90.7 | 67 | 12 | 20/04/2007 | CP000680.1 | Unpublished |
| P. putida | |||||||||
| F1 | 5 959 964 | 61.9 | 5423 | 89.9 | 76 | 20 | 31/05/2007 | CP000712.1 | Unpublished |
| GB-1 | 6 078 430 | 61.9 | 5515 | 90.2 | 74 | 22 | 05/02/2008 | CP000926.1 | Unpublished |
| KT2440 | 6 181 863 | 61.5 | 5481 | 87.5 | 74 | 22 | 22/01/2003 | AE015451.1 | Nelson et al. (2002) |
| W619 | 5 774 330 | 61.4 | 5292 | 89.8 | 75 | 22 | 11/03/2008 | CP000949.1 | Unpublished |
| P. stutzeri | |||||||||
| A1501 | 4 567 418 | 63.9 | 4237 | 90.3 | 61 | 13 | 20/04/2007 | CP000304.1 | Yan et al. (2008) |
| P. syringae | |||||||||
| pv. phaseolicola 1448A | 5 928 787 | 57.9 | 5436 | 86.8 | 64 | 16 | 01/08/2005 | CP000058.1 | Joardar et al. (2005a, b) |
| pv. syringae B728a | 6 093 698 | 59.2 | 5245 | 88.7 | 64 | 16 | 12/05/2005 | CP000075.1 | Feil et al. (2005) |
| pv. tomato DC3000 | 6 397 126 | 58.3 | 5721 | 85.6 | 64 | 16 | 21/08/2003 | AE016853.1 | Buell et al. (2003) |
| Species/strain | Size | % G+C | Genes | % coding | tRNA | rRNA | Released | GenBank | Reference |
| P. aeruginosa | |||||||||
| PAO1 | 6 264 404 | 66.6 | 5671 | 89.8 | 63 | 13 | 13/09/2000 | AE004091.2 | Stover et al. (2000) |
| PA7 | 6 588 339 | 66.4 | 6396 | 90.1 | 65 | 12 | 05/07/2007 | CP000744.1 | Roy et al. (2010) |
| UCBPP-PA14 | 6 537 648 | 66.3 | 5994 | 89.8 | 63 | 13 | 06/10/2006 | CP000438.1 | Lee et al. (2006) |
| LESB58 | 6 601 757 | 66.3 | 6026 | 88.9 | 67 | 13 | 24/12/2008 | FM209186.1 | Winstanley et al. (2009) |
| C3719 | 6 222 097 | 66.5 | 5696 | 86.6 | 40 | 6 | 04/01/2006 | NZ_AAKV00000000 | Mathee et al. (2008) |
| PA2192 | 6 905 121 | 66.2 | 6203 | 85.5 | 44 | 2 | 04/01/2006 | NZ_AAKW00000000 | Mathee et al. (2008) |
| P. entomophila | |||||||||
| L48 | 5 888 780 | 64.2 | 5293 | 89.8 | 78 | 22 | 10/05/2006 | CT573326.1 | Vodovar et al. (2006) |
| P. fluorescens | |||||||||
| Pf0-1 | 6 438 405 | 60.6 | 5741 | 90.0 | 73 | 19 | 07/10/2005 | CP000094.2 | Silby et al. (2009) |
| Pf-5 | 7 074 893 | 63.3 | 6144 | 88.7 | 71 | 16 | 30/06/2005 | CP000076.1 | Paulsen et al. (2005) |
| SBW25 | 6 722 539 | 60.5 | 6009 | 88.3 | 66 | 16 | 09/01/2008 | AM181176.4 | Silby et al. (2009) |
| P. mendocina | |||||||||
| ymp | 5 072 807 | 64.7 | 4730 | 90.7 | 67 | 12 | 20/04/2007 | CP000680.1 | Unpublished |
| P. putida | |||||||||
| F1 | 5 959 964 | 61.9 | 5423 | 89.9 | 76 | 20 | 31/05/2007 | CP000712.1 | Unpublished |
| GB-1 | 6 078 430 | 61.9 | 5515 | 90.2 | 74 | 22 | 05/02/2008 | CP000926.1 | Unpublished |
| KT2440 | 6 181 863 | 61.5 | 5481 | 87.5 | 74 | 22 | 22/01/2003 | AE015451.1 | Nelson et al. (2002) |
| W619 | 5 774 330 | 61.4 | 5292 | 89.8 | 75 | 22 | 11/03/2008 | CP000949.1 | Unpublished |
| P. stutzeri | |||||||||
| A1501 | 4 567 418 | 63.9 | 4237 | 90.3 | 61 | 13 | 20/04/2007 | CP000304.1 | Yan et al. (2008) |
| P. syringae | |||||||||
| pv. phaseolicola 1448A | 5 928 787 | 57.9 | 5436 | 86.8 | 64 | 16 | 01/08/2005 | CP000058.1 | Joardar et al. (2005a, b) |
| pv. syringae B728a | 6 093 698 | 59.2 | 5245 | 88.7 | 64 | 16 | 12/05/2005 | CP000075.1 | Feil et al. (2005) |
| pv. tomato DC3000 | 6 397 126 | 58.3 | 5721 | 85.6 | 64 | 16 | 21/08/2003 | AE016853.1 | Buell et al. (2003) |
Size, %G+C from http://www.Pseudomonas.com, apart from PA2192 and C3719 from Mathee et al. (2008).
Genes, % coding, and rRNA and tRNA content from IMG, apart from Pseudomonas fluorescens Pf0-1, SBW-25 and Pf-5 (Silby et al., 2009).
‡ The genomes of these strains have not been properly deposited in GenBank.
Coding sequences from plasmids included in gene count and calculation of % coding.
General features from completed Pseudomonas genomes
| Species/strain | Size | % G+C | Genes | % coding | tRNA | rRNA | Released | GenBank | Reference |
| P. aeruginosa | |||||||||
| PAO1 | 6 264 404 | 66.6 | 5671 | 89.8 | 63 | 13 | 13/09/2000 | AE004091.2 | Stover et al. (2000) |
| PA7 | 6 588 339 | 66.4 | 6396 | 90.1 | 65 | 12 | 05/07/2007 | CP000744.1 | Roy et al. (2010) |
| UCBPP-PA14 | 6 537 648 | 66.3 | 5994 | 89.8 | 63 | 13 | 06/10/2006 | CP000438.1 | Lee et al. (2006) |
| LESB58 | 6 601 757 | 66.3 | 6026 | 88.9 | 67 | 13 | 24/12/2008 | FM209186.1 | Winstanley et al. (2009) |
| C3719 | 6 222 097 | 66.5 | 5696 | 86.6 | 40 | 6 | 04/01/2006 | NZ_AAKV00000000 | Mathee et al. (2008) |
| PA2192 | 6 905 121 | 66.2 | 6203 | 85.5 | 44 | 2 | 04/01/2006 | NZ_AAKW00000000 | Mathee et al. (2008) |
| P. entomophila | |||||||||
| L48 | 5 888 780 | 64.2 | 5293 | 89.8 | 78 | 22 | 10/05/2006 | CT573326.1 | Vodovar et al. (2006) |
| P. fluorescens | |||||||||
| Pf0-1 | 6 438 405 | 60.6 | 5741 | 90.0 | 73 | 19 | 07/10/2005 | CP000094.2 | Silby et al. (2009) |
| Pf-5 | 7 074 893 | 63.3 | 6144 | 88.7 | 71 | 16 | 30/06/2005 | CP000076.1 | Paulsen et al. (2005) |
| SBW25 | 6 722 539 | 60.5 | 6009 | 88.3 | 66 | 16 | 09/01/2008 | AM181176.4 | Silby et al. (2009) |
| P. mendocina | |||||||||
| ymp | 5 072 807 | 64.7 | 4730 | 90.7 | 67 | 12 | 20/04/2007 | CP000680.1 | Unpublished |
| P. putida | |||||||||
| F1 | 5 959 964 | 61.9 | 5423 | 89.9 | 76 | 20 | 31/05/2007 | CP000712.1 | Unpublished |
| GB-1 | 6 078 430 | 61.9 | 5515 | 90.2 | 74 | 22 | 05/02/2008 | CP000926.1 | Unpublished |
| KT2440 | 6 181 863 | 61.5 | 5481 | 87.5 | 74 | 22 | 22/01/2003 | AE015451.1 | Nelson et al. (2002) |
| W619 | 5 774 330 | 61.4 | 5292 | 89.8 | 75 | 22 | 11/03/2008 | CP000949.1 | Unpublished |
| P. stutzeri | |||||||||
| A1501 | 4 567 418 | 63.9 | 4237 | 90.3 | 61 | 13 | 20/04/2007 | CP000304.1 | Yan et al. (2008) |
| P. syringae | |||||||||
| pv. phaseolicola 1448A | 5 928 787 | 57.9 | 5436 | 86.8 | 64 | 16 | 01/08/2005 | CP000058.1 | Joardar et al. (2005a, b) |
| pv. syringae B728a | 6 093 698 | 59.2 | 5245 | 88.7 | 64 | 16 | 12/05/2005 | CP000075.1 | Feil et al. (2005) |
| pv. tomato DC3000 | 6 397 126 | 58.3 | 5721 | 85.6 | 64 | 16 | 21/08/2003 | AE016853.1 | Buell et al. (2003) |
| Species/strain | Size | % G+C | Genes | % coding | tRNA | rRNA | Released | GenBank | Reference |
| P. aeruginosa | |||||||||
| PAO1 | 6 264 404 | 66.6 | 5671 | 89.8 | 63 | 13 | 13/09/2000 | AE004091.2 | Stover et al. (2000) |
| PA7 | 6 588 339 | 66.4 | 6396 | 90.1 | 65 | 12 | 05/07/2007 | CP000744.1 | Roy et al. (2010) |
| UCBPP-PA14 | 6 537 648 | 66.3 | 5994 | 89.8 | 63 | 13 | 06/10/2006 | CP000438.1 | Lee et al. (2006) |
| LESB58 | 6 601 757 | 66.3 | 6026 | 88.9 | 67 | 13 | 24/12/2008 | FM209186.1 | Winstanley et al. (2009) |
| C3719 | 6 222 097 | 66.5 | 5696 | 86.6 | 40 | 6 | 04/01/2006 | NZ_AAKV00000000 | Mathee et al. (2008) |
| PA2192 | 6 905 121 | 66.2 | 6203 | 85.5 | 44 | 2 | 04/01/2006 | NZ_AAKW00000000 | Mathee et al. (2008) |
| P. entomophila | |||||||||
| L48 | 5 888 780 | 64.2 | 5293 | 89.8 | 78 | 22 | 10/05/2006 | CT573326.1 | Vodovar et al. (2006) |
| P. fluorescens | |||||||||
| Pf0-1 | 6 438 405 | 60.6 | 5741 | 90.0 | 73 | 19 | 07/10/2005 | CP000094.2 | Silby et al. (2009) |
| Pf-5 | 7 074 893 | 63.3 | 6144 | 88.7 | 71 | 16 | 30/06/2005 | CP000076.1 | Paulsen et al. (2005) |
| SBW25 | 6 722 539 | 60.5 | 6009 | 88.3 | 66 | 16 | 09/01/2008 | AM181176.4 | Silby et al. (2009) |
| P. mendocina | |||||||||
| ymp | 5 072 807 | 64.7 | 4730 | 90.7 | 67 | 12 | 20/04/2007 | CP000680.1 | Unpublished |
| P. putida | |||||||||
| F1 | 5 959 964 | 61.9 | 5423 | 89.9 | 76 | 20 | 31/05/2007 | CP000712.1 | Unpublished |
| GB-1 | 6 078 430 | 61.9 | 5515 | 90.2 | 74 | 22 | 05/02/2008 | CP000926.1 | Unpublished |
| KT2440 | 6 181 863 | 61.5 | 5481 | 87.5 | 74 | 22 | 22/01/2003 | AE015451.1 | Nelson et al. (2002) |
| W619 | 5 774 330 | 61.4 | 5292 | 89.8 | 75 | 22 | 11/03/2008 | CP000949.1 | Unpublished |
| P. stutzeri | |||||||||
| A1501 | 4 567 418 | 63.9 | 4237 | 90.3 | 61 | 13 | 20/04/2007 | CP000304.1 | Yan et al. (2008) |
| P. syringae | |||||||||
| pv. phaseolicola 1448A | 5 928 787 | 57.9 | 5436 | 86.8 | 64 | 16 | 01/08/2005 | CP000058.1 | Joardar et al. (2005a, b) |
| pv. syringae B728a | 6 093 698 | 59.2 | 5245 | 88.7 | 64 | 16 | 12/05/2005 | CP000075.1 | Feil et al. (2005) |
| pv. tomato DC3000 | 6 397 126 | 58.3 | 5721 | 85.6 | 64 | 16 | 21/08/2003 | AE016853.1 | Buell et al. (2003) |
Size, %G+C from http://www.Pseudomonas.com, apart from PA2192 and C3719 from Mathee et al. (2008).
Genes, % coding, and rRNA and tRNA content from IMG, apart from Pseudomonas fluorescens Pf0-1, SBW-25 and Pf-5 (Silby et al., 2009).
‡ The genomes of these strains have not been properly deposited in GenBank.
Coding sequences from plasmids included in gene count and calculation of % coding.
Data retrieved 23 December 2010.
Only Pseudomonas spp. for which more than one strain has been sequenced are shown.
‡ Number of genes in a particular isolate not present in other members of the same species.
In all cases, data were derived from the Joint Genome Institute IMG system (http://img.jgi.doe.gov/), using maximum E-value=1e-5, and minimum % identity=30.
Data retrieved 23 December 2010.
Only Pseudomonas spp. for which more than one strain has been sequenced are shown.
‡ Number of genes in a particular isolate not present in other members of the same species.
In all cases, data were derived from the Joint Genome Institute IMG system (http://img.jgi.doe.gov/), using maximum E-value=1e-5, and minimum % identity=30.
By comparison with the genome sequences of P. aeruginosa PAO1 and P. syringae, the nonpathogenic lifestyle of P. putida was correlated with the absence of key animal and plant pathogenicity genes, i.e. KT2440 did not harbour genes for a type III protein secretion system (T3SS), exotoxin A, alkaline protease, elastase, a rhamnolipid biosynthesis operon, exolipase or phospholipase C, or genes specifying plant cell wall-degrading enzymes found in plant pathogens. The presence of genes with roles in disease such as quorum sensing (QS), alginate biosynthesis and adhesins in KT2440, albeit with some modifications, supports a broader role of these genes in the ecological success in addition to their known contributions to the virulence of P. aeruginosa.
Pseudomonas putida strains live in diverse soil environments. Analysis of the KT2440 genome uncovered features reflecting the adaptability of P. putida and their applicability to bioremediation (Nelson et al., 2002; Weinel et al., 2002). Pseudomonas putida KT2440 encodes 350 outer membrane and cytoplasmic transporters (15% more than PAO1) for the uptake/efflux of a wide range of substrates. Also identified were genes predicted to be involved in the transformation of a range of aromatic compounds not previously known to be substrates of KT2440 metabolic pathways, suggesting that even broader applications of the species in bioremediation may be possible. A xenB homologue, involved in 2,4,6-trinitrotoluene catabolism, and 12 glutathione S-transferases, predicted to be involved in detoxification, were also identified. KT2440 has 11 LysE family amino acid efflux transporters, compared with one in PAO1, indicating that P. putida may need to reduce the levels of amino acids in the cell to avoid them becoming inhibitory to growth or that the transporters serve to transport other as yet unknown molecules.
Using the KT2440 genome sequence and a microarray, the similarity among P. putida strains for which genome sequences were not available was analysed (Ballerstedt et al., 2007). Based on the presence/absence of genes, strains were grouped into two clusters, one of which included a Pseudomonas monteilii strain previously thought to be distal to the P. putida group.
Pseudomonas fluorescens– biocontrol and plant growth-promoting bacteria
Pseudomonas fluorescens are generally regarded as plant commensals and have been studied for their biocontrol and plant growth promotion properties (Haas & Defago, 2005). Pseudomonas fluorescens genomes have a high number of genes with no known function (Tables 1 and 2) (Paulsen et al., 2005; Silby et al., 2009). Among the three sequenced P. fluorescens strains, there is a conserved set of 3642 CDSs, and 1490, 1400 and 1111 CDSs found in Pf-5, SBW25 and Pf0-1, but not in the other two strains (Silby et al., 2009). Much of the P. fluorescens genomes are asyntenous, indicating a large degree of rearrangement. Remarkably, P. fluorescens Pf-5 was found to have a very high number (1052) of gap-free 34-bp REP sequences, compared with 21 in P. aeruginosa PAO1, 365 in P. syringae DC3000 (Tobes & Pareja, 2005) or 804 in P. putida KT2440 (Aranda-Olmedo et al., 2002). REP sequences were absent from regions of the Pf-5 genome that encode housekeeping genes, reflecting the strong selection pressure for maintenance of function and the dynamically adaptive variable regions. In KT2440, the distribution of REP sequences was heavily biased toward intergenic spaces, possibly reflecting similar selection pressure (Aranda-Olmedo et al., 2002). The genomes of Pf-5, SBW25 and Pf0-1 also carry intergenic repeat sequences distinct from the REP sequences, which can be categorized into 12 families ranging in size from 50 to 352 bp (Silby et al., 2009). The distribution and density of these repeats vary considerably among the genomes. The density in SBW25 was sufficiently high to allow the detection of a nonrandom distribution around the genome, whereby so-called ‘repeat deserts’ lacking repeat sequences were found to cover 40% of the genome. The functional significance of REP and other repeats is not clear, but their presence might provide a substrate for recombination events that rearrange existing genes and incorporate newly acquired ones into the genome.
Like P. putida KT2440, the saprophytic lifestyle of P. fluorescens in soil and on plants is reflected in the genome sequence: the three genomes lack genes for most known virulence factors, although SBW25 carries genes for a T3SS similar to those found in the pathogenic Pseudomonas species. Despite the fact that some T3SS genes known to be important in P. syringae type III secretion are absent, the SBW25 system is expressed in the sugar beet rhizosphere and is capable of secretion (Rainey et al., 1999b; Preston et al., 2001; Jackson et al., 2005). No plant disease has been associated with the SBW25 T3SS, consistent with the genome-informed assessment that SBW25 harbours a low complement of plant-targeting putative type III secreted effectors (T3SE). SBW25 does, however, harbour effector ExoY (Vinatzer et al., 2005) (found in P. aeruginosa), which is thought to target the actin cytoskeleton of eukaryotic cells (Engel & Balachandran, 2009). The T3SS of other P. fluorescens strains have been implicated in haemolysis (Sperandio et al., 2010) and for targeting the oomycete pathogen Pythium ultimum (Rezzonico et al., 2005). This may indicate that SBW25 uses the system against an alternative host. It is also possible that SBW25 represents a ‘snapshot’ of a strain in the process of losing or acquiring the system (Jackson et al., 2005).
Genomic analysis has provided an insight into the environmental success of P. fluorescens. Each genome encodes a wide array of membrane-based sensors and receptors as well as enzymes for diverse catabolic activities, including breakdown of plant-derived substrates such as lignin (a trait of P. putida). Pf-5 is well known as a producer of secondary metabolites with antimicrobial activity, such as the polyketides pyoluteorin (Howell & Stipanovic, 1980) and 2,4-diacetylphloroglucinol (Nowak-Thompson et al., 1994), which may be vital for the bacterium to fend off competitors in the soil and plant environment, and are also key targets for exploitation in the biological control of plant diseases (Weller et al., 2007). Examination of the Pf-5 genome sequence suggested sequences that may specify previously undetected toxins (Paulsen et al., 2005). By focusing on sequences that resemble nonribosomal peptide synthetases or polyketide synthases, a cyclic lipopeptide with activity against oomycete zoospores was discovered (Gross et al., 2007). Loper and colleagues used sequence-guided targeted mutagenesis to functionally analyse a Pf-5 gene cluster (rzx) predicted to be responsible for making analogues of the antifungal/anti-oomycete rhizoxin secondary metabolite (Loper et al., 2008). Comparative metabolic profiling of wild type and rzx mutant strains confirmed the genome-based prediction of rhizoxin synthesis. Five rhizoxin analogues were described, one of which was novel, and were shown to have varied toxicity to different plants and nonspecific antitumour activity with human tumour cell lines. The potential use of these newly discovered compounds for biotechnology is obvious, but their contribution to ecological success is less clear. If they play a role in competitive fitness, their unique occurrence in Pf-5 among the P. fluorescens isolates suggests a niche-specific activity that may be accomplished by different mechanisms in other isolates.
A genome-wide functional screen for environmentally induced loci (EIL) in SBW25 identified 125 sequences that show elevated expression in planta (Silby et al., 2009). Putative orthologues of 73 of these sequences occur in both Pf0-1 and Pf-5, indicating that environmental responses (and presumably adaptation) are mediated by both conserved and strain-specific factors. The functions of SBW25 genes induced in planta and shared among all three strains include motility, nutrient scavenging, stress response, detoxification and regulation, each of which would be predicted to be important in natural environments. Genome-wide screens for regulators of these EIL identified several known and novel regulators, including seven regulators controlling the expression of a cellulose synthase gene system known to be important for plant colonization (Gal et al., 2003; Giddens et al., 2007). Intriguingly, several ‘reverse orientation’ sequences were also induced in planta in SBW25, suggesting the existence of microRNAs and protein-coding antisense genes similar to cosA and other antisense genes in Pf0-1, which are difficult to predict in genomes (Silby & Levy, 2004, 2008; Kim et al., 2009).
Pseudomonas stutzeri– a nitrogen-fixing nonfluorescent pseudomonad
Pseudomonas stutzeri A1501 survives in soil and colonizes plant roots epiphytically and endophytically (Lalucat et al., 2006). Consistent with its saprophytic lifestyle, pathogenicity and virulence genes, including those for T3SS and T4SS, were not found in the genome (Yan et al., 2008). The ability to fix nitrogen means the bacterium has been developed as a bioinoculant to stimulate plant growth. The P. stutzeri genome is small compared with other Pseudomonas genomes at 4.56 Mbp, encoding just 4146 predicted proteins, highlighting the considerable genomic variation that has been noted in the genus (Tables 1 and 2). The genome lacks intact prophages, but does contain 42 repeat sequences and 57 transposases. Four potential GIs were identified based on an atypical nucleotide composition and association with tRNA genes. Comparison of protein products with the predicted proteomes of other Pseudomonas species revealed the closest relatedness to P. aeruginosa, and analysis of the conserved gene complement among 14 Pseudomonas spp. indicates that P. stutzeri is somewhat distantly related to the other sequenced Pseudomonas spp. (Silby et al., 2009), and shares approximately 56% of protein-coding genes with the nitrogen-fixing bacterium A. vinelandii strain DJ (Setubal et al., 2009).
Genes most likely to be required for nitrogen fixation and rhizosphere competence were identified in a 49-kb region containing 59 genes in P. stutzeri A1501, which, based on the G+C content, was postulated to be a GI acquired through horizontal transfer, and inserted between cobS (PST_1301) and PST_1360 (Yan et al., 2008). The high level of gene synteny in the flanking regions in other Pseudomonas species, coupled with variability between those genes, leads to the suggestion that this is a hot spot for insertion or variation. Other factors likely to contribute to rhizosphere competence include >300 transporter genes (87 unique to P. stutzeri), several of which appear to serve for uptake of dicarboxylic acids, carbohydrates and amino acids that are commonly found in plant root exudates. The genome also revealed gene systems likely to be important for success in fluctuating soil environments such as those for the degradation of aromatic compounds, nitrate metabolism, osmotolerance, detoxification of reactive oxygen species, chemotaxis, type IV secretion (for DNA transformation) and extracellular polysaccharide (cellulose) production. A gene for inhibiting ethylene production was also found, which could contribute to plant growth promotion by reducing ethylene inhibition of root development. Pseudomonas stutzeri does not appear to harbour known siderophore biosynthesis genes, consistent with its description as nonfluorescent. So how do P. stutzeri survive in iron-limiting environments? The genome sequence indicates the presence of three putative siderophore receptors (PST_0147, 0996 and 4174). Pseudomonas stutzeri may act as a cheat, utilizing siderophores produced by other pseudomonads (Loper & Henkels, 1999; Harrison & Buckling, 2009). Alternatively, P. stutzeri may have alternative iron acquisition systems: our analysis of the genome sequence found that four putative siderophore biosynthesis protein genes are present in the genome, three of which are clustered (PST_2504, 3699, 3701-2). If these were involved in siderophore production, they clearly do not confer a fluorescent phenotype.
Pathogenicity
Some pseudomonads cause disease in diverse host organisms. The most studied and economically important species are the opportunistic pathogen, P. aeruginosa, and the plant pathogen, P. syringae. The economic and health costs associated with Pseudomonas infections have driven an intense interest in their genome sequences. Comparative genomic approaches show the diversity of genes among pathogenic isolates, highlighting the extent of host–pathogen coevolution and the range of mechanisms potentially used to cause disease in different hosts.
Pseudomonas aeruginosa– opportunistic human pathogen
Pseudomonas aeruginosa is an aquatic and soil bacterium that can infect a range of organisms, including plants (Rahme et al., 2000), nematodes (Tan et al., 1999a, b), fruit flies (Apidianakis & Rahme, 2009), waxmoth (Miyata et al., 2003), zebrafish (Clatworthy et al., 2009) and various mammals (Snouwaert et al., 1992; Colledge et al., 1995; Potvin et al., 2003). In humans, P. aeruginosa is an opportunistic pathogen capable of infecting a range of tissues and sites (Lyczak et al., 2000) and is one of the top six infectious disease threats (Talbot et al., 2006), causing serious infections in patients who are immunocompromised or whose natural defences are otherwise breached. For example, it is associated with infections of burns and wounds patients, and infections of the eye associated with contact lens use (Robertson & Cavanagh, 2008). Pseudomonas aeruginosa is also the major pathogen contributing to the morbidity and mortality associated with cystic fibrosis (CF), causing chronic lung infections, and makes a considerable contribution to the burden of hospital-acquired infections (Govan & Deretic, 1996), where it is a frequent cause of respiratory and urinary tract infections, a common cause of hospital-acquired pneumonia, healthcare-associated pneumonia and ventilator-associated pneumonia (American Thoracic Society, 2005). Pseudomonas aeruginosa infections are particularly challenging because of the organisms' broad intrinsic antimicrobial resistance (Lister et al., 2009).
Pseudomonas aeruginosa is a challenging pathogen to combat due to the formation of a sessile biofilm during persistent infections (Costerton et al., 1999), which enhances resistance to antimicrobial treatments and to host immune defences (Costerton & Lewandowski, 1995; Drenkard & Ausubel, 2002; Mah et al., 2003). The involvement of three overlapping QS systems (Whiteley et al., 1999; Girard & Bloemberg, 2008), a T3SS (Hauser et al., 2009), sigma factors (Potvin et al., 2008) and two-component regulatory systems in controlling virulence and resistance in P. aeruginosa (Gooderham & Hancock, 2009) suggests that numerous virulence factors controlled by these systems may be revealed by functional genomic analysis.
PAO1 encodes a large number of regulatory (521), outer membrane (∼150) and cytoplasmic membrane proteins (∼300), which is consistent with its exposure to diverse environments. The bacterium encodes a number of transporters putatively involved in antibiotic resistance, highlighting the significant problems associated with controlling P. aeruginosa infections. Genes for T1SS, T2SS and T3SS as well as a number of genes for chemotaxis and motility were also identified. In the original annotation of the PAO1 genome, 2549 (46% of genome) genes were originally annotated to be of unknown function or homologous to genes with unknown function (some of these were reclassified; Weinel et al., 2003). This fact almost certainly illustrates the diversity of genes that contribute to the complex ecology of the bacterium. In November 2010, 2445 genes were still classified as having an unknown function, indicating that with the benefit of 10 years of functional research and comparative genomics, only around 100 unknown genes have been functionally characterized.
The size of the PAO1 genome results from genetic complexity (more genes of unique function) rather than gene duplications. Pseudomonas aeruginosa has a large number of paralogous groups (distinct gene families), indicating that its genome has evolved through genetic expansion (Stover et al., 2000), which may suggest adaptation to colonization of and competitive fitness in a diverse range of ecological niches. The function of many genes may become apparent only when analysed within more complex environments than generally used in laboratories, as has been argued in the case of environmentally induced genes in P. fluorescens (Rainey et al., 1999b; Silby & Levy, 2004).
As a means to determine the genomic basis for varied degrees of virulence in different P. aeruginosa isolates, a PAO1 array was constructed and used to evaluate the presence and absence of virulence genes in 18 strains of environmental or clinical origin (Wolfgang et al., 2003). The virulence gene data failed to correlate strongly with an associated phenotypic appraisal of the virulence properties of the 18 isolates. Clearly, the combination of variable genes in each genome has a considerable impact on pathogenesis. When used in conjunction with a functional approach (a comprehensive transposon mutant library screen), the genome sequence did aid in the identification of common P. aeruginosa virulence factors as well as PA14-specific virulence genes, but it is striking that Lee et al. (2006) concluded that it may not be possible to predict the virulence (or otherwise) of one strain based on the presence (or absence) of genes known to be important for the pathogenicity of a different strain.
Comparative analysis of P. aeruginosa genomes
After PAO1, the genome of P. aeruginosa PA14 was sequenced. PA14 generally shows greater virulence than PAO1 and thus provided an opportunity to further explore the genetic basis for virulence in this species. Ninety-two per cent of the PA14 genome is present in PAO1 and 96% of PAO1 in PA14. The PA14 genome harbours 322 more CDSs than PAO1; strain-specific genes typically cluster together in areas of the chromosome exhibiting a low GC content relative to the rest of the chromosome i.e. on probable GIs. In all, 58 PA14-specific gene clusters (with 478 genes) and 54 PAO1-specific gene clusters (with 234 genes) were identified. The major virulence-related genomic differences between PA14 and PAO1 are the presence of two pathogenicity islands in PA14 (He et al., 2004). Remarkably, 63% of the PA14-specific CDSs have no known function compared with 29% for the genome on the whole, suggesting PA14-specific adaptations to an unknown condition, by uncharacterized mechanisms.
It is clear that GIs and prophages contribute most to variations among the genomes of P. aeruginosa strains. The genome sequences of three P. aeruginosa strains, PA2192, PACS2 and C3719, isolated from CF patients have been analysed (Table 1) (Mathee et al., 2008). PA2192 exhibits major phenotypic adaptation including conversion to mucoidy while C3719, a representative of the Manchester Epidemic Strain (Jones et al., 2002), is known for its transmissibility. A comparison of five P. aeruginosa genomes (PAO1, PA14, PACS2, PA2192 and C3719) revealed a relatively large set of 5021 conserved genes, in contrast to the modest conserved complement seen in comparative analysis of P. fluorescens strains (Silby et al., 2009). Pseudomonas aeruginosa genomes are organized as mosaics of conserved regions interspersed by insertions containing blocks of strain-variable genes found in a limited number of chromosomal locations termed ‘regions of genomic plasticity’ (RGP) (Mathee et al., 2008). Some of these RGPs are unique to a strain because of the deletion of the gene(s) in other strains, but many of the strain-specific RGPs have been horizontally acquired. Importantly, the islands inserted at RGPs often differ among genomes, indicating the individual evolutionary trajectories of the strains, while also maintaining core virulence functions. Although there are 11 named P. aeruginosa GIs (PAGI1-11) (Battle et al., 2009) in addition to PAI-1 and PAPI-2, it is clear that with each new genome sequenced, new GIs can be added to the list.
The P. aeruginosa Liverpool Epidemic Strain (LES) is a particularly successful and aggressive transmissible strain associated with lung infections in CF patients. The genome of the earliest isolated LES genotype, LESB58, was sequenced with the aim of understanding the genetic basis of its high virulence and its emergence as a significant pathogen (Winstanley et al., 2009). The LESB58 genome harbours all except two of the 265 PAO1 virulence genes. However, significant variations were identified, including duplications of pyoverdine-associated genes and divergence in gene complement and homology of type IV pili and phenazine biosynthesis genes. Importantly, specific mutations could be correlated to phenotypic changes. For example, LESB58 is nonmotile and analysis of the genome identified a pseudogene form of gene PA1628, which had previously been found to be important for motility and is thus is likely to be responsible for this phenotype. Reduced motility in CF isolates is not uncommon (Starkey et al., 2009), although the causes of this adaptation are not always known. A mutation in a pseudogene of the GDP-mannose-4,6-hydratase gene could be responsible for the rough colony morphotype of the O-antigen-negative LESB58. It is of considerable importance to realize that sufficient genomic and functional data now exist to allow sequence changes to be correlated with phenotypic differences.
The major difference between the genome of LESB58 and other P. aeruginosa genomes was the presence of five GIs and six prophages, some of which were novel, while others were related to similar elements in other sequenced strains. Signature-tagged mutagenesis (STM) demonstrated that genes located in a GI (LES GI-5) and prophages (LES prophages 1, 2, 3, 5) were essential for the competitiveness of this strain in a rat model of chronic lung infections (Winstanley et al., 2009), showing that the acquisition of unique variable sequences can enhance success, and emphasizing the role of bacteriophages in the evolution of virulence.
The genome sequence of the clinical (nonrespiratory, multiresistant) taxonomic outlier P. aeruginosa PA7 revealed considerable divergence from other P. aeruginosa strains (Roy et al., 2010). In syntenic regions, the PA7 genome has only 93.5% identity with other sequenced strains (PAO1, PA14, LESB58), which generally show 99.5% identity (Spencer et al., 2003). The genome has a similar representation of functional categories of genes, except for a bias towards more DNA replication, repair and recombination genes due to a high number of transposase and integrase genes in GIs. Of the 51 GIs identified, 18 are novel to PA7. Among these islands were genes for heavy metal efflux, ectoine utilization, iron transport, haemagglutinins, fimbrial genes and multidrug efflux genes, representing acquired adaptive traits. The genome sequence was particularly useful for unravelling the genetic basis of antibiotic resistance, at which PA7 excels: point mutations in gyrA and parC confer fluoroquinolone resistance and efflux systems with conserved and unique (oprA) components were the primary modes of resistance. Intriguingly, the PA7 genome lacks the genes for the structural T3SS as well as effectors exoS, exoU and exoY, and several other known virulence genes were absent. Despite the importance of T3SS in some pathogenic P. aeruginosa isolates, it cannot be considered part of the conserved core genome of P. aeruginosa. Strains generally carry the T3SS genes necessary for the secretion of either exoS or exoU, and absence of the structural T3SS genes has only been reported for PA7. The PA7 genome also highlighted the limitations of using certain genes in multilocus sequence typing (MLST) analyses, with only two of seven commonly used genes showing enough conservation to be useful. PA7 is clearly an atypical P. aeruginosa, and some would suggest that it may represent a different species altogether.
The Pseudomonas genome database (http://v2.pseudomonas.com) has played an important role in maintaining updated information via community-based sequence annotation and more recently providing valuable tools for comparative genomics (Winsor et al., 2009).
Functional genomics
Transposon mutagenesis is a mainstay of bacterial genetic research. In combination with complete genome sequences, additional power is gained by enabling the precise mapping of numerous insertion points, and their relative positions in the genome. Both Jacobs et al. (2003) and Lewenza et al. (2005) created saturated transposon mutant libraries in PAO1. After sequencing and mapping insertion points, Jacobs and colleagues finished with a defined library of 30 100 unique insertions (five hits per CDS, excluding 678 CDSs probably encoding essential functions). Similarly, 20 530 unique transposon insertions were mapped within the PA14 genome (Liberati et al., 2006). Mutants of PAO1 (http://pseudomutant.pseudomonas.com/) and PA14 (http://ausubellab.mgh.harvard.edu/cgi-bin/pa14/home.cgi) are available to the research community. A ‘track’ in GBrowse on the http://v2.pseudomonas.com website facilitates easy identification of orthologues of disrupted genes in other sequenced Pseudomonas spp. Dotsch et al. (2009a) used the PA14 mutant library to identify genes involved in antibiotic resistance, and suggest that data from this functional genomic approach will allow the rapid prediction of resistance phenotypes of clinical isolates for which some DNA sequence is obtained.
STM is a modification of transposon mutagenesis that can be used to identify genes that are important during infections. Using a rat model for chronic lung infections, STM was coupled with genome sequence data as a high-throughput screening tool to identify virulence genes (Potvin et al., 2003). This initial study, using PAO1, has since been extended to identify in vivo essential genes in the CF Liverpool Epidemic Strain (LESB58) (Winstanley et al., 2009). Of the 47 STM mutants essential for competitiveness in a rat model of chronic lung infection, three mapped to different LES prophages and one to a novel GI, demonstrating the importance of the accessory genome in the pathogenicity of this strain. A similar approach has been used to identify genes that are important for P. aeruginosa pathogenicity in Caenorhabditis elegans and mice (Wiehlmann et al., 2007a).
Numerous studies have exploited the development of microarrays, designed based on genome sequence data, in order to study gene expression in response to different environments, such as during biofilm growth (Whiteley et al., 2001) or exposure to oxidative stress (Salunkhe et al., 2005). Dotsch et al. (2009b) created a custom-made Affymetrix tiling array of 250 000 oligonucleotides of PAO1, covering 85% of the genome to identify single nucleotide polymorphisms (SNPs) and insertion/deletion events in PAO1 and PA14 genomes. It was used to identify mutations in naturally occurring PAO1 mutants that exhibit β-lactam resistance and upregulation of a cepahlosporinase AmpC (Moya et al., 2009).
Pseudomonas syringae– plant pathogen
Since P. syringae was first described, similar bacteria have been isolated from numerous plants showing disease symptoms. Although once numbering >40 species, they are now classified as the single species P. syringae sensu lato (Palleroni et al., 1984): this general classification reflects the lack of adequate taxonomic studies to properly classify the strains. Indeed, DNA similarity indicates that P. syringae is a very diverse group and should be considered as a species complex. Depending on their interactions with given plants, P. syringae strains are assigned to one of over 50 pathogenic variants (pathovars; pvs) (Dye et al., 1980). For example, P. syringae pv. tomato (Pto) infects Solanum lycopersicum L. (tomato). Studies of P. syringae have identified several complex networks of interactions between plant defence mechanisms and pathogen-associated molecular patterns (Jones & Dangl, 2006). The T3SS, common to all pathogenic strains, is essential for facilitating the injection of multiple effector proteins into plant cells to interact with protein targets (suppress plant innate immune defences, manipulate hormone signalling and/or elicit cell death) (Cunnac et al., 2009). The genomes of multiple P. syringae pathovars have been, or are being sequenced, with a major goal to use genomic methods to characterize T3SS effector repertoires and other features that could contribute to host range and specificity (Lindeberg et al., 2008). Pseudomonas syringae also produces phytotoxins during the host–pathogen interaction that generally lack host specificity and can directly injure cells of both host and nonhost plants (Bender et al., 1999).
Pto strain DC3000 is a model P. syringae strain because it also infects Arabidopsis thaliana, a model plant for development, physiology and resistance studies. The Pto DC3000 genome (Table 1) includes two plasmids of 73 and 67 kb (Buell et al., 2003). The large number of genes of unknown function (Buell et al., 2003) highlights how little is known about this species, and the probable complexity of its life under natural environmental conditions. Unlike P. aeruginosa and P. putida, Pto DC3000 exhibits a relatively high level of gene duplication, with 2735 genes (48%) in 687 paralogous families.
New genes pertinent to life in plants were discovered in the DC3000 genome. A γ-aminobutyric acid (GABA) permease gene, three GABA transaminase genes and a succinate semialdehyde dehydrogenase gene were found and predicted to be involved in the utilization of GABA, which is abundant within the apoplast of tomato (Rico & Preston, 2008). A second important observation was that Pto DC3000 has a higher number of sugar transporters, but a lower number of amino acid transporters than P. aeruginosa PAO1 and P. putida KT2440 (Buell et al., 2003). Several of the sugar transporters are predicted to transfer plant-derived sugars such as xylose, arabinose and ribose. These two observations showed new aspects of the P. syringae lifestyle and point to a degree of semi-specialism linked to the pathogen's ecology. In the DC3000 genome, 298 genes were categorized as virulence factors broadly encompassing type III protein secretion, siderophores, phytotoxins, adhesins, extracellular polysaccharides, pectic enzymes and detoxification of antimicrobials such as reactive oxygen species and copper, of which PAO1 has 191. Sixty-five genes were unique to DC3000, and 33 of these were predicted to be type III effectors or helpers. This large complement of effectors explains the functional redundancy inherent to the T3SS and reflects multiple rounds of pathogen evolution to overcome host resistance (Stavrinides et al., 2006). The absence of genes for the production of known lipodepsinonapeptide phytotoxins such as syringomycin or syringopeptin, which can contribute to virulence (Scholz-Schroeder et al., 2001), hints at mechanistic heterogeneity beyond T3SEs.
The genome of a second P. syringae pv. tomato strain, T1, which, unlike DC3000, cannot infect Arabidopsis, aligns with DC3000 over most of its length. However, 757 CDSs are unique to T1, of which 167 are putative virulence genes (Almeida et al., 2009). Differences in the T3SE content were apparent, with some unique in Pto T1 while some found in DC3000 were absent. The T1 genome provides an opportunity to unravel the mechanisms of host range between two very closely related pathogens.
The P. syringae B728a and 1448A genome sequences have provided an insight into two bean pathogens: the brown spot pathogen P. syringae pv. syringae (Psy) B728a and the halo blight pathogen P. syringae pv. phaseolicola (Pph) 1448A (Feil et al., 2005; Joardar et al., 2005a). The Psy B728a genome is smaller than that of DC3000, and shares 4273 genes with DC3000 (Feil et al., 2005). At least 375 REP sequences were identified in B728a (cf. 365 in DC3000; 1052 in P. fluorescens Pf-5; 21 in P. aeruginosa), but only 16 insertion sequence (IS) elements were found and unlike DC3000 and 1448A, the strain does not carry a plasmid. Nine major genomic rearrangements exist between DC3000 and B728a. Many of the novel genes in B728a were on one of 14 GIs, one of which is an integrative and conjugative element (ICEland) similar to that found in Pph 1302A and P. fluorescens Pf-5 (Paulsen et al., 2005; Pitman et al., 2005).
Although the Pph 1448A genome sequence is smaller than B728a (5.92 Mbp), it is more densely coded, with more CDSs spread between one chromosome and two plasmids (Joardar et al., 2005a). Like Pto DC3000, the largest class of genes in Pph 1448A was categorized as transporters (14%) and >500 (10%) genes were predicted to encode regulators, emphasizing the ecological and genetic complexity of P. syringae. Between 1448A and DC3000, 77% of CDSs were syntenic. The identification of 1348 paralogous families is indicative of a high level of gene duplication in the genome, similar to DC3000.
Psy B728a causes brown spot disease of bean leaves and has been studied extensively in its epiphytic and infectious phases of life (Hirano et al., 1999; Monier & Lindow, 2003, 2004, 2005). Consistent with its life on the relatively barren surface of the leaf, a wide range of genes important for ecological success were predicted and indeed identified, including iron-scavenging siderophores, UV resistance, QS, copper and antibiotic resistance, osmotolerance and ice nucleation (Marco et al., 2005). Genes likely to be important for virulence after apoplast infection included enzymes to target plant tissue (cellulose, pectate lyase, xylanase), auxin biosynthesis genes, two toxins (syringomycin and syringopeptin) and several polyketide/nonribosomal peptide synthases to make unknown products. The genome sequence also revealed 27 putative T3SEs, of which five were not found in DC3000 (Feil et al., 2005). Of these, 22 were proven to be secreted by the T3SS (Vinatzer et al., 2006).
Of the 298 virulence-related CDSs identified in Pto DC3000 (Buell et al., 2003), 81% are shared with 1448A, and 1448A encodes at least 22 experimentally verified T3SEs (Chang et al., 2005) and potentially up to 34 (Studholme et al., 2009); 18 are shared with Pto DC3000. One unusual feature of the 1448A genome is the presence of two T3SS, the second of which is probably not involved in plant pathogenicity (Lindgren et al., 1986; Mansfield et al., 1994; Joardar et al., 2005a). Because the closest orthologues are in P. aeruginosa and Photorhabdus, the system may play a role in interactions with different eukaryotic hosts such as insects, nematodes or amoeba. Also of interest in the Pph 1448A genome was the presence of two T2SS, whereas Pto DC3000 only carries one system: 1448A has more candidate substrates (e.g. cell wall-degrading enzymes) for secretion than DC3000 (Joardar et al., 2005a). Pph 1448A also encodes three nonribosomal peptide synthase gene clusters including the phaseolotoxin gene cluster, the toxin responsible for causing the chlorotic haloes at sites of infection, but lacks coronatine toxin genes. Altogether, the 1448A genome highlights the relative similarity between Pto and Pph, but also identified novel regions that may be attributable to host specificity.
Draft genomes of P. syringae pathovars
Reinhardt et al. (2009) produced a draft of the rice pathogen P. syringae pv. oryzae (Por) genome. Por is highly diverged from the other P. syringae strains, with only 41±8% identity between the Por and the Pto genomes. The Por genome sequence allowed whittling of the conserved set of genes in P. syringae to 3594 CDSs; 97 genes were found to be unique to Por. The complement of T3SEs (29 effectors were predicted) was found to be unique compared with other P. syringae genomes, possibly facilitating a major goal of P. syringae genomics, determination of the extent to which these gene complements define plant host range.
The tobacco pathogen P. syringae pv. tabaci (Pta) genome carries 303 CDSs not present in Pph 1448A, Psy B728a or Pto DC3000, and is most closely related to Pph 1448A (97% identity) (Studholme et al., 2009). The 24 T3SE genes identified enabled the reappraisal of the core effector complement to just 12 genes common to all P. syringae pathovars. Other differences of possible importance for niche specialization between Pta and Pph were that Pta lacked polysaccharide-modifying enzymes and Rhs insecticidal toxins, but did carry a tabtoxin cluster that is not present in Pph. A total of 102 regions of >1 kb were identified as putative Pta GIs. Pta revealed a wide range of differences among P. syringae pathovars outside of the effector complement that may point the way forward in the analysis of adaptations to specific hosts.
Recent drafts of the genomes of P. syringae pathogens of tree hosts [P. syringae pv. savastanoi (Psv) NCPPB 3335 (also known as P. savastanoi pv. savastanoi; Gardan et al., 1992) and P. syringae pv. aesculi (Pae)] have provided a rapid insight into the potential mechanisms behind their ability to infect woody tissues (Green et al., 2010; Rodríguez-Palenzuela et al., 2010). The tumour-inducing olive tree pathogen Psv causes olive knot disease. The genome carries 73 genes unique to Psv, and has eight major inversions, with two postulated to be in Pph after comparison between Pph, Psv and Psy B728a (Rodríguez-Palenzuela et al., 2010). The 73 Psv-specific genes included those for a range of membrane proteins, transporters and regulators. Also discovered were 12 variable regions, many of which had the features of GIs. One of these contains a region of genes predicted to be involved in the utilization of anthranilate as the sole carbon and nitrogen source and for the degradation of catechol. Other genes probably encode proteins for the breakdown of lignin-related aromatic compounds, such as vanillin, to protocatechuate; genes for protocatechuate metabolism were also detected, suggesting that the pathogen has evolved a system enabling the breakdown of woody tissue of the olive tree. Although 11 genes specifying novel T3SEs were found in Psv, these may not be central to the host specificity. Rather, the combination of capabilities conferred by the 73 Psv-specific genes may be the critical factors for providing Psv access to an otherwise impenetrable host.
The United Kingdom and Western Europe are currently experiencing an epidemic of bleeding canker disease, caused by Pae, in European Horse Chestnut trees (Aesculus hippocastanum) (Webber et al., 2008; Green et al., 2010). Phylogenetic comparison found the Pae strains grouped with other pathovars of woody hosts, including Psv and P. syringae pv. morsprunorum (causes canker in cherry trees). Three genome sequences of European (E-Pae) isolates and the Indian type strain (I-Pae) that causes leaf spot symptoms in Aesculus indica were sequenced to study the evolution of the E-Pae strains and to identify the causes for their sudden emergence in the early 2000s (Green et al., 2010). The E-Pae and I-Pae genomes are 95% similar, with about 300 kb and 1613 SNPs differing among them. The E-Pae strains, isolated from different geographic regions of the British Isles, varied by only three nucleotides and in their plasmid content, suggesting that the E-Pae strains share a recent ancestor and were likely derived from a single introduction to Britain.
The Pae genomes all harbour a T3SS and have an effector content similar to that of Pph 1448A (Green et al., 2010). They also carry putative lignin-degradation genes similar to those found in Psv, reinforcing the hypothesis that these systems are important for infection of woody host plants. Pae isolates carried various haemolysin and filamentous-haemagglutinin genes, as well as xaxAB insect toxin genes, which could indicate an association with insect vectors (Vigneux et al., 2007). Intriguingly, the Pae strains harbour a cluster of enterobactin siderophore biosynthesis genes; these are normally found in enteric bacterial pathogens. This is the first discovery of the enterobactin siderophore in a pseudomonad, complementing production by P. syringae of the more common pyoverdine and the less widespread yersiniabactin (Bultreys et al., 2006; Jones et al., 2007a; Cornelis et al., 2010). Because enterobactin is the most powerful iron-binding siderophore known, it may indicate that the host tree, or an alternative host, is particularly deficient in available iron. E-Pae strains harbour sucrose utilization genes, which are absent from I-Pae, reflecting the pathology of the two pathogens; E-Pae strains grow in the sucrose-rich vascular systems of tree stems and branches, while I-Pae is found in leaves, where sucrose is more scarce.
The genome of the nonpathogenic P. syringae Psy642 does not carry the typical T3SS or the conserved and exchangeable effector loci flanking the hrp/hrc genes (Clarke et al., 2010). However, Psy642 and other phylogenetically similar nonpathogenic strains can still colonize leaf surfaces and endophytic spaces of plants. Of interest, Psy642 encodes at least seven putative effectors including AvrE, ExoY and ExoU homologues, and an atypical T3SS in a different genomic location. Two genes (hrpK and hrpS) were absent from the cluster. This T3SS is capable of secreting effectors into plant cells, but the gene cluster was not necessary for growth on plants. There is some parallel with the T3SS of P. fluorescens SBW25, i.e. both look like restructured T3SS capable of effector secretion, but lack hrpS, have limited effectors including avrE and exoY and are not essential for plant colonization. These secretion systems may serve an alternative purpose, perhaps in interactions with nonhost plants or insects (Clarke et al., 2010).
Dissection of virulence in P. syringae
Bioinformatic analysis of P. syringae genomes has helped in the identification of secretion system genes and their substrates, which aids in the identification of putative virulence factors such as the T3SS and T3SEs (Fouts et al., 2002). Two transcriptomic studies complemented these analyses using a DC3000 oligo-based array (Lan et al., 2006) or an ORF-based array (Ferreira et al., 2006) to identify genes regulated by the T3SS regulators HrpRS and HrpL (the enhancer binding proteins HrpRS activate hrpL and the alternate transcription factor HrpL binds to hrp box promoters to activate T3SS structural genes and effectors). The HrpL regulon generally overlapped the HrpRS regulon, but a number of HrpL-independent genes were identified, indicating the more global effect of HrpRS (Lan et al., 2006). As well as finding known or predicted T3SS regulated genes, other genes not predicted through bioinformatics were identified. Interestingly, housekeeping and flagellum biosynthesis genes were downregulated when T3SS genes were upregulated, which is probably an adaptive response similar to that seen previously (Wolfgang et al., 2004) to prevent triggering of basal immunity in the plant host (Zipfel et al., 2004). Ferreira et al. (2006) found 119 genes upregulated (which is in good agreement with Lan et al., 2006) and 76 genes downregulated by HrpL. This analysis helped to identify the full regulon controlled by the T3SS regulator as well as gain a better understanding of the HrpL cis promoter sequence. In a follow-up experiment, the Lon protease regulon was analysed (Lan et al., 2007) as Lon is known to lead to the degradation of HrpR and T3SE proteins (Bretz et al., 2002; Losada & Hutcheson, 2005), and is important for full virulence of DC3000 in tomato leaves (Lan et al., 2007). In the DC3000 lon mutant, 155 genes were upregulated including hrpL and 39 genes in the HrpL regulon, while 48 genes were downregulated, including genes for cell membrane lipoproteins, extracellular polysaccharides and lipopolysaccharide. The differential levels of expression of HrpL-regulated genes in the lon mutant indicate that additional regulatory signals affect their expression.
Pseudomonas entomophila– insect pathogen
Pseudomonas entomophila was isolated from fruit flies and was subsequently found to be a pathogen of Drosophila (Vodovar et al., 2005). The taxonomic standing of P. entomophila is unclear, as the species has not been described formally (Mulet et al., 2010). Based on the genome sequence (Tables 1 and 2) (Vodovar et al., 2006), the closest relative to P. entomophila is P. putida, with 70% of P. entomophila genes present in P. putida: 96% of these are in synteny. The difference in the remaining genes is largely attributed to a high number of paralogous genes in P. putida, rather than a reduction in genome size in P. entomophila leading to gene loss. Like several other members of the genus, P. entomophila carries numerous REPs of unknown function, distributed widely in the genome (Vodovar et al., 2006).
Because of the interest in P. entomophila as a model for insect pathogenesis and host–pathogen interactions (Vodovar et al., 2005), genome analysis was focused on the identification of processes that might contribute to virulence. The gene complement of P. entomophila includes genes that may specify proteins required for survival, persistence and immune avoidance in insect hosts and systems to produce insecticidal toxins including three TccC-type toxins similar to those of Photorhabdus luminescens (ffrench-Constant & Waterfield, 2005) and more distantly related TccC- and TcdB-type toxins. Also identified were a putative repeats-in-toxin (RTX) gene and a T1SS unique to P. entomophila, which may be responsible for the pathogen's haemolytic activity (Vodovar et al., 2005). Four lipases, three serine proteases and an alkaline protease represent other potentially secreted biologically active proteins that could contribute to virulence. Unusually for a Gram-negative pathogen, P. entomophila does not encode a T3SS or T4SS; this is particularly interesting, given that the ‘specialist’ insect pathogen P. luminescens harbours a T3SS important for avoiding phagocytosis (Brugirard-Ricaud et al., 2005), as do several strains of the opportunist P. aeruginosa. Although isolation of P. entomophila from a noninsect source has yet to be reported, we suggest that the abundance of transporter and regulator genes (535 and >300, respectively) reflects the probable ecology of the pathogen – two lifestyles, one in insects and the other in the environment. This suggestion is in line with the lifestyles of other Pseudomonas pathogens.
Adaptation in environmental and host niches
Many virulence determinants in P. aeruginosa are generally viewed as being a part of the conserved gene set (Lee et al., 2006; Mathee et al., 2008), in contrast to P. syringae, where effectors are often found linked to mobile DNA such as transposons, GIs and plasmids (accessory loci). It has been suggested that the content of the accessory genome in P. aeruginosa determines environmental adaptability such as niche expansion (Mathee et al., 2008). However, accessory genome regions can contribute to virulence (He et al., 2004) or fitness to establish infections (Winstanley et al., 2009). Thus, these differences may reflect two facts: (1) P. aeruginosa is a more homogeneous pathogen that has emerged relatively recently compared with the P. syringae species complex and (2) P. aeruginosa is a versatile and flexible opportunist capable of causing infections in diverse hosts and at multiple sites within hosts, while P. syringae pathovars are more specialized. Accessory genes may contribute to the increased virulence or competitiveness of particular strains of P. aeruginosa in specific niches.
Diversity in gene content
While the majority of genes have probable orthologues in other members of the same species, there are a large number of unique protein-coding genes in Pseudomonas genomes when compared with closely related isolates (Table 2). Among P. aeruginosa isolates, Mathee et al. (2008) determined that there is a conserved set of 5021 genes, but between P. aeruginosa isolates, a substantial variation can be seen. For example, PA7 has 660 CDSs that are absent from other sequenced P. aeruginosa isolates (maximum E-value=1e-5; minimum % ID=30; Table 2). Two experimental approaches that analysed clone libraries from collections of clinical strains foreshadow what might be expected when large numbers of P. aeruginosa isolates are sequenced. Shen et al. (2006) generated a large clone genomic library from 12 strains of P. aeruginosa from which 348 (approximately 11%) of a set of randomly chosen clones harboured sequences distinct from those found in PAO1. In a similar approach, 1300 random clones from a large library derived from 12 clinical P. aeruginosa strains were examined; 13% contained sequences not found in the PAO1 genome or GenBank (Erdos et al., 2006). There is clearly a greater degree of variation among P. aeruginosa isolates than generally considered, which will become more apparent with increased numbers of sequenced genomes. In an interesting counterpoint to the variability that may exist among P. aeruginosa isolates, Dotsch et al. (2010) found that among 36 clinical isolates, there are 980 genes for which there was absolute conservation at the protein sequence level, and yet only 124 of those could be classified as essential based on mutant studies.
A considerable degree of genetic diversity is apparent in other Pseudomonas species for which more than one isolate has been sequenced. For example, P. syringae pv. tabaci 11528 has 575 CDSs that have no orthologues in P. syringae pv. phaseolicola (most closely related strain) (Studholme et al., 2009). Among the three P. fluorescens strains sequenced to date, only 3642 genes were conserved (Silby et al., 2009), and each isolate carried a large number of CDSs unique to the species (Table 2). The genome-specific genes, whether in GIs or not, extend the concept of adaptability and flexibility commonly attributed to pseudomonads. In addition to the unifying trait of large genomes with a broad repertoire of functional abilities, different isolates have adapted to their particular niches by expanding the pan-genome.
Inferring lineage-specific adaptation from genome sequences
The ratio of nonsynonymous to synonymous site substitutions (dN/dS) has been used to evaluate whether positive, relaxed or purifying selection is the predominant selective force on genome evolution (Yang & Bielawski, 2000). The more sequences that are available, the more in-depth such analyses. Shapiro & Alm (2009) introduced an additional means by which to measure patterns of selection, based on the ratio of ‘slow’ vs. ‘fast’ substitutions (S: F); it finds deviations from a sequence's ‘normal’ selection regime. Using these methods, lineage-specific adaptations that might have escaped detection because of the absence of appropriate experimental data may be revealed, opening new areas of investigation.
The S: F ratio is potentially informative when applied to a group of closely related isolates for which whole-genome sequences are available. An elevated S: F at a particular locus in one branch of a lineage can point to a specific, possibly ecologically relevant, adaptation that the majority of organisms in the lineage has not undergone. S: F is less useful when several members of a lineage are under similar selective pressures, because it would not be possible to classify particular sites as generally ‘slow’ changing if indeed there is a high degree of variability at those sites. When applied to Pseudomonas genomes, Shapiro & Alm (2009) detected an elevated S: F ratio in some energy production proteins [pyruvate dehydrogenase E1 component (AceE) and succinate dehydrogenase complex (SdhCD)], which in most species tend to have low S: F ratios. Although S: F alone cannot discern between positive or relaxed negative selection when a lineage such as Pseudomonas AceE and SdhCD show a high S: F compared with other lineages, it does potentially indicate ecological adaptation and thus allow for the generation of testable hypotheses. For example, Shapiro & Alm (2009) proposed a possible link between the changes in P. fluorescens with growth in oxygen-limiting biofilms, which has led to the selection of phenazine compounds as terminal electron acceptors. Moreover, changes in SdhCD may reflect changes in redox balance associated with oxidative stress experienced by Pseudomonas interacting with eukaryotes such as plants. Whether these data indicate positive selection for an adaptive change or a relaxed negative selection remains to be determined.
Genomic Islands, bacteriophage and plasmids – the accessory genome in Pseudomonas spp.
Bacteriophages
Although it seems likely that there is a finite number of phage types in the P. aeruginosa pan-genome, there is often sequence variation even between related prophages in different genomes (Ceyssens & Lavigne, 2010), with prophage genomes often exhibiting a mosaic structure. Among the six phage-related gene clusters in P. aeruginosa strain LES (isolate LESB58 with five complete and one defective phage), only the gene cluster encoding a Pf1-like filamentous prophage is shared with PAO1 (Winstanley et al., 2009). LES prophages 2 and 3 share a duplicated region with each other, share some regions with a previously sequenced phage (F10) and contain novel regions. LES prophage 4 is highly related, but not identical to the previously sequenced phage D3112, while LES prophage 5 contains some regions similar to another characterized phage, D3, and also shares a duplicated region with LES prophage 3. Hence, there is evidence for duplications, genetic rearrangement and recombination driving diversity in prophages, and the emergence of LES isolates with different combinations of prophages suggests that prophages contribute to genome instability in LES during infection (Fothergill et al., 2010b).
GIs
GIs, encompassing pathogenicity islands and fitness islands, are frequently associated with genes that contribute to pathogenicity or fitness (Arnold et al., 2003; Battle et al., 2009; Jackson et al., 2011). In P. aeruginosa LESB58, >75 genes were found on three novel islands. Interestingly, the 110-kb LESGI-3 island showed a bipartite structure, with 67 kb identical to PAGI-2, indicating significant island evolution. One of the novel islands harboured the biosynthetic genes for the antifungal pyoluteorin, found in some P. fluorescens strains, while the other two carried a mix of regulator, transporter, sensor and restriction–modification genes (Winstanley et al., 2009).
Variants of the plasmid/integrative and conjugative element (ICE) pKLC102 have been identified in P. syringae B728a and several P. aeruginosa strains. Pseudomonas aeruginosa clone C isolates from the lungs of CF patients and from aquatic environments (Romling et al., 1994) carry variants of pKLC102, often integrated as an island, with sequence characteristics indicating both plasmid and phage origins (Klockgether et al., 2004). Genome sequencing has revealed related elements in P. aeruginosa strains PA14, PA7, 2192, C3719, LES and PACS2, but not PAO1. In all sequenced pKLC102-like elements, there are homologues of 75–98 of the 103 genes in pKLC102, of which a conserved set of 34 are probably involved in transfer, replication and recombination (Würdemann & Tümmler, 2007). Island-specific genes make up the remainder of pKLC102-like GIs. In PA14, the pKLC102-like GI is the pathogenicity island PAPI-1. The relatedness of strains carrying pKLC102-like elements does not correlate with the relatedness of the elements themselves (Würdemann & Tümmler, 2007), indicating frequent transfer, a possibility that is well supported by experimental evidence of transfer (Qiu et al., 2006; Lovell et al., 2009) and by the diversity and broad distribution of such elements (Klockgether et al., 2007).
In P. fluorescens strains, genome sequences have revealed the diversity of integrated elements. In Pf-5, nine GIs, some of which encode secondary metabolites, and six phage-like regions were identified, some resembling phages found in the enteric Shigella and Yersinia (Mavrodi et al., 2009). The search for GIs uncovered insertion hotspots, such as between the mutS and cinA genes, where P. fluorescens strains have prophage-like elements (Mavrodi et al., 2009; Silby et al., 2009), and P. putida GB-1, P. syringae DC3000 and P. entomophila have various other genes. A 115-kb ICE island (ICEland) PFGI-1 was found inserted into a tRNALYS gene on the Pf-5 chromosome similar to an ICEland found in P. syringae (Jackson et al., 2000; Pitman et al., 2005), and related elements are present in SBW25 and Pf0-1 (Silby et al., 2009), revealing their ubiquity as well as the heterogeneity of gene content carried in the cargo regions of the ICElands (Reva et al., 2006; Mavrodi et al., 2009). A P. syringae ICEland (PPHGI-1) carrying a T3SE can excise from the P. syringae chromosome during growth within resistant plants and transfer to recipient strains by transformation or conjugation. This can also lead to other genomic changes in the recipient, demonstrating the impact that GIs can have on the evolution of new pathogen types (Pitman et al., 2005; Lovell et al., 2009, 2010; Godfrey et al., 2010, 2011).
Plasmids
Autonomously replicating plasmids that do not integrate into the host genome have so far been uncommon in the sequenced Pseudomonas spp. P. syringae isolates DC3000 and 1448A both carry two plasmids, but the plasmids in one strain are quite distinct from those in the other (Joardar et al., 2005a). Several virulence factor homologues are present on the plasmids p1448A-A and p1448A-B, but are found on the DC3000 chromosome. Similarly, most of the virulence factor genes on DC3000 plasmids are chromosomal in 1448A. The virulence factors on p1448A-A include the gene virPphA (hopAB1) essential for virulence in bean cultivars and an avirulence gene recognized by soybean (Jackson et al., 1999, 2002). However, the PAI lacks avrPphF (hopF1) due to transposase-mediated rearrangement (Tsiamis et al., 2000; Rivas et al., 2005). The virulence genes on the DC3000 plasmid pDC3000A have chromosomal paralogues, raising the possibility that they are not essential for the pathogenicity of DC3000 (Buell et al., 2003). The plasmid pQBR103 (and a variety of other plasmids) was identified in P. fluorescens SBW25 after the previously plasmid-free SBW25 was introduced into sugar beet crops as a seed dressing (Tett et al., 2007). Although the original isolate of SBW25 did not carry a plasmid (Silby et al., 2009), the acquisition of plasmids such as pQBR103, in which only 20% of CDSs are associated with functional predictions, demonstrated that there is a considerable pool of elements moving among environmental bacteria in the phytosphere.
Lineage-specific regions (LSR) and Regions of Genomic Plasticity (RGP)
Joardar et al. (2005b) compared the genomes of DC3000 with PAO1 and KT2440 to identify LSRs, many of which were hypothesized to be involved in fitness or pathogenicity. This was done by identifying LSRs and features within them associated with horizontal gene transfer such as an altered G+C content and an atypical nucleotide composition. In total, 44 LSRs were identified in DC3000, of which 34 are larger than 10 kb. Of the 983 CDSs within LSRs, 533 are unique to DC3000. Many CDSs with an atypical trinucleotide composition or an altered G+C content were found in the LSRs, indicating a correlation between LSRs and lateral gene transfer. Interestingly, parts of the DC3000 chromosome are enriched with LSRs and parts appear to be preferred sites for genes acquired by horizontal transfer; in particular, the surE-mutS region is associated with a chromosomal inversion likely catalysed by homologous recombination between ISPsy6 insertion sequences. In P. putida, the surE-mutS region contains an intron-encoding maturase, another indication that this could be a hotspot for insertion of mobile DNA.
Based on their CDS content, the 44 P. syringae DC3000 LSRs were grouped into phage, fitness, virulence, extrachromosomal and MGEs, and unknown. The virulence LSRs include the hrp T3SS pathogenicity island, the coronatine toxin cluster and islands containing T3SE genes. Several of these were inserted into tRNA genes, including the 99-kb LSR10, which is a pKLC102-like element related in part to the PAPI-1 island of P. aeruginosa and PPHGI-1 of P. syringae pv. phaseolicola (He et al., 2004; Pitman et al., 2005). Interestingly, analysis of T3SEs and hrp box promoters (part of the type III regulon) found 83% and 85%, respectively, associated with LSRs. One of the phage LSRs also contains T3SEs. The fitness LSRs contained genes for sucrose utilization, cyanate detoxification, resistance to tellurite and macrolides, chemotaxis genes and a nitrilase. Although nitrilase activity has not been detected in Pto DC3000, a Psy B728a nitrilase enables the utilization of indole-3-acetonitrile and phenylpropionitrile as nitrogen sources, and also mediates synthesis of the plant hormone indole acetic acid by the bacterium, which may favour the bacterium–plant interaction (Howden et al., 2009). Overall, this study highlighted the power of comparative genomics to identify species-specific genes that may otherwise have been overlooked through traditional approaches. In strain 1448A, almost 5% (256) of CDSs are related to MGEs. Several transposase genes are present, the majority of which differ between the B728a and 1448A genomes, indicating that they were acquired after divergence of the two lineages.
In their comparison of five P. aeruginosa genomes, Mathee et al. (2008) identified 52 RGPs that can result either from insertions of GIs or phage genomes or from the deletion of genes from one or more strains. Using subtractive hybridization, Battle et al. (2008, 2009) identified seven putative GIs (termed PAGI-5–11) in the highly virulent P. aeruginosa PSE9 that were absent from PAO1. Only three of those GIs were correlated with RGPs, and the gene content of the islands was different from that found in five sequenced isolates (Mathee et al., 2008), indicating that knowledge of P. aeruginosa GIs is not yet complete in terms of both the possible genomic locations and the range of cargo genes carried.
Adaptation in the CF lung
Pseudomonas aeruginosa is the most common pathogen infecting the lungs of CF patients, causing chronic infections that contribute to the morbidity and mortality associated with the disease. In contrast to their behaviour in acute infections, in chronic infections, P. aeruginosa populations diversify into multiple coexisting phenotypic variants. These manifest themselves in a number of ways, including the emergence of colony morphology variants (Govan & Deretic, 1996; Haussler et al., 2003), hypermutability (Oliver et al., 2000) and diversity in both antimicrobial susceptibility and virulence traits (von Gotz et al., 2004; Fothergill et al., 2007). Using genomics to study sequential isolates, it was demonstrated that specific mutations often occur during this process of adaptation, and this has led to the suggestion that loss of virulence may be advantageous (Smith et al., 2006). However, diversity in P. aeruginosa populations is a striking feature of chronic infections, and some members of the population maintain virulence capabilities even after many years of infection (Fothergill et al., 2010a).
Genome organization
Variation in the distribution of ‘conserved’ genes
Not only does gene content vary among isolates, but at another level – the genome-wide distribution of orthologous genes – the diversity and variability of Pseudomonas genomes are apparent. There is considerable flexibility in the genomes of pseudomonads, evidenced by the occurrence of large inversions and translocations such as those observed during long-term infections by P. aeruginosa (Ernst et al., 2003; Kresse et al., 2003). A striking example is the inversion of a 1.7-Mb segment of the sequenced PAO1 genome relative to the PAO1 sibling isolate DSM-1707 (Schmidt et al., 1996, 1998), which occurred in a research laboratory sometime after 1990, and that is also apparent when comparing PAO1 with PA14 (Fig. 2), resulting in the replication terminus being moved to a position 38.8% of the way around the circular chromosome with respect to the origin (Lee et al., 2006). Additional diversification of the PAO1 genome has been detected recently using genome resequencing of sublines derived from the original PAO1 isolate. The changes include duplication of a prophage region, deletions and SNPs (Klockgether et al., 2010), revealing microevolution in PAO1 and highlighting the plasticity of the genome. A similar inversion phenomenon was seen for the P. syringae pv. tomato DC3000 genome, which was attributed to transposon and plasmid variation (Landgraf et al., 2006), and resequencing of the Psy B728a genome identified 11 nucleotide substitutions that may represent errors in the original sequence or mutations that have arisen in the samples (Farrer et al., 2009). These studies highlight the importance of maintaining isogenic stocks and the speed with which substantial genomic changes can accumulate as bacteria adapt to their laboratory environments.
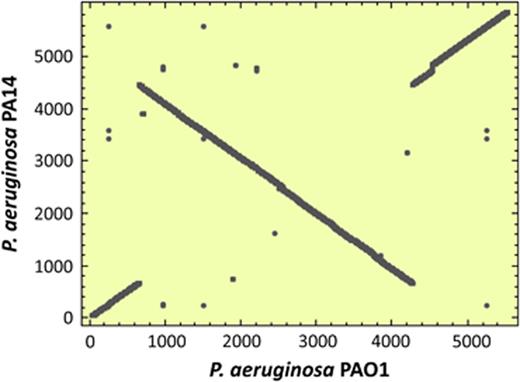
Geneplot (http://www.ncbi.nlm.nih.gov/sutils/geneplot.cgi) comparisons showing pairs of orthologous genes for Pseudomonas aeruginosa PAO1 and PA14. Each axis shows the number of predicted proteins. Note the large inversion, which reflects the reported inversion in the PAO1 genome (Stover et al., 2000).
When comparing the three completely sequenced P. fluorescens isolates, extensive reciprocal recombination about the replication terminus was detected (Silby et al., 2009). In addition, some loci are inverted in one strain relative to the others, and numerous other loci have been translocated, with or without also being inverted (Fig. 3). The sequences least susceptible to such a rearrangement are found close to the origin of replication. Similarly, among P. aeruginosa isolates, there is a high degree of stability around the replication origin, with more rearrangements of small genomic regions apparent closer to the replication terminus, although it should be noted that the actual sequences into which RGPs are inserted are highly conserved (Mathee et al., 2008). Alignment of complete P. syringae genomes indicates translocations and inversions similar to those seen in P. fluorescens, although there is less recombination about the replication terminus. Related groups of Pseudomonas spp. such as P. fluorescens and P. syringae appear to be very tolerant of genetic rearrangement. The underlying processes mediating these rearrangements are not always clear. Numerous repetitive sequences, which could be substrates for homologous recombination, have been identified in P. fluorescens in addition to the rRNA operons (Paulsen et al., 2005; Silby et al., 2009), but the boundaries of many rearrangements do not map to those repeat sequences.

Comparison of the complete six-frame translations of the whole-genome sequences of Pseudomonas fluorescens Pf0-1, SBW25 and Pf-5. The analysis was carried out using the artemis Comparison Tool and computed using tblastx. Forward and reverse strands of DNA are shown for each genome (dark grey lines). The red bars between the DNA lines represent individual tblastx matches, with inverted matches coloured blue. Note the inversions and translocations, and the reduced number of orthologous pairs near the replication terminus (center of figure).
In the genomes of P. fluorescens isolates, the synteny and proportion of orthologous genes are high near the origin of replication, but decrease toward the replication terminus. This split is not easily explained, and does not correlate with prophages or ‘atypical’ regions in P. fluorescens genomes (Silby et al., 2009) (Figs 3 and 4). Highly expressed genes may cluster near the replication origin because of increased gene dosage during replication resulting from increased copy number. Conversely, poorly expressed genes, of which horizontally transferred genes are often examples, may cluster nearer the terminus (Rocha, 2004). The latter point would be consistent with the regions of low orthology. However, support for bias toward the preferential accumulation of mobile genes at the terminus is weak (Rocha, 2004), and there are few indicators in P. fluorescens that the terminus region has widespread incorporation of mobile elements (Silby et al., 2009).
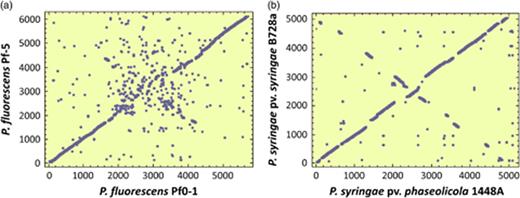
Geneplot comparisons showing pairs of orthologous genes for (a) Pseudomonas fluorescens Pf-5 and Pf0-1 and (b) Pseudomonas syringae B728a and 1448A. The axes show the number of predicted proteins. Close to the origin of replication, there is a high degree of synteny in both species. Note the deterioration of synteny in P. fluorescens genomes near the replication terminus, compared with the P. syringae isolates, which show a high degree of synteny along with a few inversions.
Genome-based taxonomy of Pseudomonas
There are at least two goals for classifying microbial species. One is to categorize organisms together based on similarity, allowing predictions of behaviour. A second goal is to use the relationships among strains and species in an evolutionary context, to understand ancestral life and the processes that have shaped life as we know it. In short, the questions are where did they come from and what do they do now? With knowledge of evolution potential, it may be possible to predict the outcomes of projected evolution, for example how a pathogen evolves antibiotic resistance.
In the absence of morphological traits and breeding range barriers that have aided the taxonomic classification/species concept in multicellular sexual creatures, classification systems for bacteria have traditionally depended on phenotypic and biochemical properties, and more recently on small-scale, but informative molecular information. The power of the latter is exemplified by the revolutionary reclassification of life into three kingdoms based on rRNA sequences (Woese & Fox, 1977). The advent of genome-based methods provides an opportunity to examine the relatedness of bacteria at an unprecedented level of molecular detail.
Even at a quick glance, questions regarding the classification of pseudomonads into species groups arise. It is common for a given isolate of a particular ‘species’ to have a gene complement in which >8% of the genes have no orthologues in other members of the ‘species’. While some of this variation may be accounted for by horizontal gene flow, and thus dismissed as ‘accessory’ information, it is unlikely that all of those genes are the product of recent transfer events. In the case of P. fluorescens, there are a large number of unique genes in each strain, but there is little evidence that the majority of these were recently acquired, pointing to a long-term evolutionary process and highlighting that P. fluorescens is probably a species complex (Silby et al., 2009).
Mathee et al. (2008) presented an inferred phylogenetic relationship generated from concatenating 1836 ORFs that are common to P. fluorescens and six strains of P. aeruginosa, showing that the multihost pathogen PA14 falls outside the main P. aeruginosa cluster. Within the main cluster, the relative distance between branch nodes also suggests significant evolutionary divergence among the remaining P. aeruginosa strains. It was suggested that this phylogenetic tree for the six P. aeruginosa strains is in agreement with their phenotypes and origins, and that PA14 and PA2192 may be phylogenetic outliers due to their larger genome sizes (Mathee et al., 2008); however, we consider this unlikely, given that the analysis was restricted to shared orthologous loci.
Mathee et al. (2008) also used a maximum parsimony method to infer phylogenetic relatedness based on concatenation of conserved genes. Comparing this tree with individual gene trees led to the selection of a list of 144 phylogenetically useful genes within the species that approximate the whole genome-based phylogeny. Importantly, this list lacks most of the genes commonly used in phylogenetic analyses including MLST, highlighting the potential improvements that can be generated using genome-based analyses (see supporting information in Mathee et al., 2008). The utility of the list for the broader genus remains to be determined, and should be tested using the 240 strains of diverse origin, which Wiehlmann et al. (2007b) show form two major and multiple minor clonal groups.
Bacterial species definition is a controversial subject (Staley et al., 2006). Mutations, inversions and acquisitions all act to muddle the various analyses and the borderline between species is particularly murky. DNA sequence analysis has helped to clarify phylogenetic relationships with MLST and multilocus sequence analysis (MLSA) providing strong discrimination. MLST uses four to 10 gene sequences and alleles are assigned numbers. If two strains have the same alleles for the gene set being used, then they are assigned to one sequence type (ST), whereas differences result in the assignment of unique STs; analysis of populations is then based on ST numbers. Conversely, MLSA uses the actual DNA sequences for phylogenetic analysis and it is considered more powerful for analysis of bacteria with less clearly defined species boundaries (Gevers et al., 2005; Almeida et al., 2010). Guttman et al. (2008) argue that MLST data based on housekeeping genes provide a strong phylogenetic signal allowing accurate construction of evolutionary relationships among pseudomonads. This may be true, given the relatively low frequency of recombination observed for housekeeping genes, but gene selection will have a major influence on the outcome because some genes are in linkage disequilibrium while other core genes appear to be freely recombining (Wiehlmann et al., 2007a). Genome sequences provide access to all housekeeping genes, and with the increased availability of draft genome sequences, it is probable that robust evolutionary models can be constructed. Constructing MLST-based trees based on genome sequence data will provide a key resource for assessing newly isolated pseudomonads. The growing number of genome sequences also makes it possible to sample more genes for their reliability in phylogenetic profiling, providing guidance to the Pseudomonas community, as was done with Escherichia coli (Konstantinidis et al., 2006). Average nucleotide identity (ANI) and average amino acid identity (AAI) use pairwise comparisons of isolates based on genes and proteins conserved between particular pairs to determine relationships. Using MLST, ANI and AAI, and a gene-independent analysis of dinucleotide composition, recent studies have directly examined the relatedness of different Pseudomonas spp (van Passel et al., 2006; Goris et al., 2007; Silby et al., 2009; Mulet et al., 2010). Collectively, these studies indicate that several species of Pseudomonas are close to or meet the current expectations of a bacterial species. However, in the case of P. syringae and P. fluorescens, the situation is less clear. Pseudomonas syringae sensu stricto refers to P. syringae pv. syringae, but the species designation has also been applied as a provisional solution to encompass all pathovars of the pathogen (Dye et al., 1980). Low DDH values between P. syringae strains led to the definition of nine genomospecies (Gardan et al., 1999). Moreover, it was shown that Pto DC3000 and Psy B728a had low DDH, ANI and AAI values below the species thresholds and therefore may not be part of the same species (Fig. 5, Goris et al., 2007; Konstantinidis & Tiedje, 2007), suggesting that strains classified as P. syringae should be regarded as part of a P. syringae species complex.
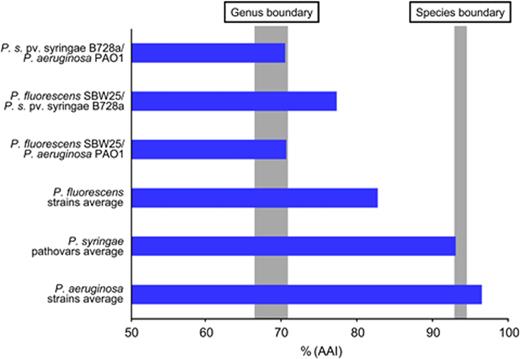
AAI for pairs of Pseudomonas spp. While Pseudomonas fluorescens strains do not reach the species cutoff, they are more closely related than SBW25 is to PAO1. Genus and species boundaries are as defined (Konstantinidis & Tiedje, 2007).
Pseudomonas fluorescens
Pseudomonas fluorescens is a diverse group of pseudomonads and has been split into various biovars based on phenotypes and biochemical activity (Stanier et al., 1966). Genomic analyses are now confirming these data, and providing a molecular viewpoint on the basis for this variation. The MLSA analysis by Guttman et al. (2008) using gapA, gltA, gyrB and rpoD demonstrated that the P. fluorescens clade is very diverse, and actually includes several other species. There was also lower confidence in the branching within the P. fluorescens clade than in the other clades, as has been seen elsewhere (Yamamoto et al., 2000; Silby et al., 2009). Pseudomonas fluorescens trees have similar topology whether derived from the shared genome content (Silby et al., 2009), eight housekeeping genes (gyrB, rpoB, rpoA, recA, gyrA, rpoD, gltA and gapA) (Rodríguez-Palenzuela et al., 2010), four housekeeping genes (16S rRNA gene, gyrB, rpoB and rpoD) (Mulet et al., 2010) or two genes (gyrB and rpoD) (Yamamoto et al., 2000).
In terms of overall relatedness, genomic approaches have demonstrated the variability among P. fluorescens isolates. The number of orthologous genes identified as reciprocal best blast hits was unexpectedly low, totalling just 59.3% of CDSs in strain Pf-5, and concentrated in 2/3 of the genome around the replication origin (Fig. 3). Unique genes comprised 22–27% of the CDSs in P. fluorescens (Silby et al., 2009). This is in contrast to P. aeruginosa, which showed that depending on the strain, the P. aeruginosa isolates had a percentage of unique genes ranging from 1.4% to 8.2% (Mathee et al., 2008). The P. aeruginosa genome sequences all come from strains isolated from clinical settings, raising the possibility that strain choice may bias our view of the genome.
Using dinucleotide frequency as a measure of the genome signature, van Passel et al. (2006) demonstrated that the genus Pseudomonas has a slightly above average genomic dissimilarity score, indicating a slightly greater than average variation within the genus. The dissimilarity score between P. fluorescens Pf0-1 and Pf-5 genomes was extremely high, suggesting that they might eventually be categorized as separate species.
Analysis of three completely sequenced P. fluorescens strains using DDH and ANI (Goris et al., 2007) and AAI and a phylogenomic approach (Silby et al., 2009) has been particularly revealing. By any of the measures applied, it could be argued that the three P. fluorescens strains should not be considered as one species. The AAI for P. fluorescens was approximately 83% (Silby et al., 2009), which is well below the species cutoff (Konstantinidis & Tiedje, 2005). The AAI for other Pseudomonas spp. gets closer to or above the species cutoff, although some P. putida strains fall slightly below the species cutoff limit (Fig. 6). However, it is not clear whether these data are sufficient to reclassify P. fluorescens into different species groups. Phylogenomic analysis indicates that the P. fluorescens strains are more closely related to each other than they are to other members of the genus, but the different P. fluorescens strains do not reach the species cutoff when compared (Figs 5 and 7).
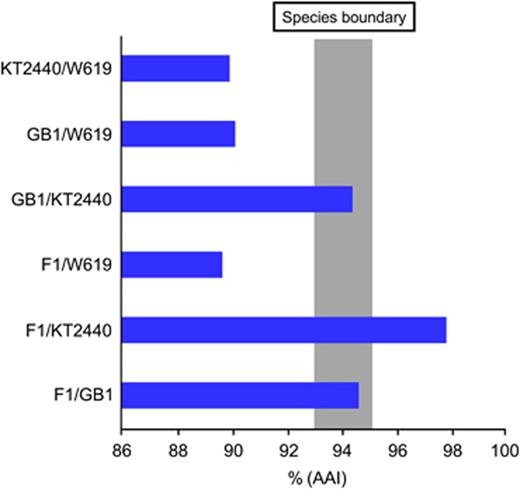
AAI for pairs of Pseudomonas putida isolates. Species boundary is as defined by Konstantinidis & Tiedje (2007). Note that some pairings fail to reach the species cutoff. KT2440, W619, GB1 and F1 are P. putida strains (see Table 1 for details).
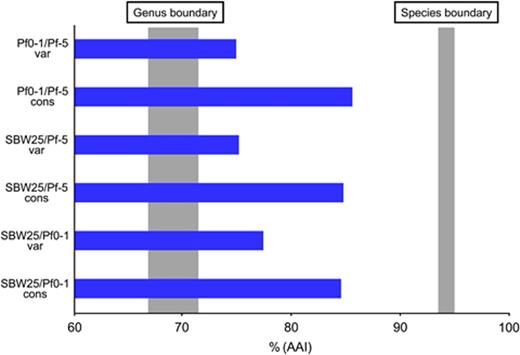
Comparison of conserved (cons) and variable (var) regions in pairs of Pseudomonas fluorescens isolates, as measured by average amino acid. Note that the AAI is considerably lower for conserved sequences in the more variable region than for those sequences in the conserved portion of the P. fluorescens genomes. Genus and species boundaries are as defined (Konstantinidis & Tiedje, 2007).
It is clear that the evolutionary relationships can be well established using MLST, and ANI/AAI are powerful methods for establishing pairwise relationships. Such techniques allow major goals of taxonomy to be fulfilled – understanding the evolutionary history of an organism, and the systematic ordering of those organisms. However, the fact that these methods necessarily must ignore more variable (or accessory) portions of the genome, where several phenotypes of interest are encoded, means that a second goal, that of functional predictability, cannot always be fully met.
Concluding comments
There is much that can be learned from the analysis of the linear sequence of nucleotides, when appropriate tools are applied. Carrying out comparative analyses adds further depth to the data, and provides the opportunity to make predictions about the ecological causes for differences in gene complement among related strains and species. The gene predictions from the initial publications are continually being revised, and it should be noted that different informatic approaches yield different conclusions [e.g. P. fluorescens Pf-5 in http://pseudomonas.com (6145 CDSs) and in IMG (6142 CDSs)]. It seems clear that further sequence data will aid in building a better picture of the genus Pseudomonas– in fact, judging by the breadth of mutations and horizontal acquisitions found in many of the described studies, it is evident that the full nature of genomic diversity is far from being realized. With each new completed genome, the estimated conserved gene complement of the genus is revised downward. The conserved complement in five P. aeruginosa isolates was 5021 genes, but when a P. fluorescens strain was included in the analysis, the useable set of genes for phylogenetic analysis declined to 1836 (Mathee et al., 2008). In an analysis of 14 Pseudomonas genomes, the common gene set was reduced to 1705 genes (Silby et al., 2009).
After 15 years of microbial genome sequencing and analysis, and 10 years of Pseudomonas genome sequencing, we have barely scratched the surface of a complex microbial world – many of the hypothetical genes of the species are still a mystery. How do scientists address this issue? We suggest that something as crucial as expression data and phenoarray (metabolomic) analyses (Jones et al., 2007b; Rico & Preston, 2008) should be sought as a matter of priority to verify transcriptional activity and identify the signals and environments that trigger expression. Although the emergence of new and relatively inexpensive sequencing technology has already accelerated the rate at which Pseudomonas genome sequences are being determined, these technologies are not being rapidly used for postgenomic analyses.
The use of high-throughput transcriptomic and proteomic technologies should routinely follow genome sequencing to provide a level of experimental verification of gene predictions. Using microarrays or transcriptome sequencing, the presence of transcripts (under specified growth conditions) can be easily verified and used to support computational annotation of the genome (e.g. Yoder-Himes et al., 2009; Filiatrault et al., 2010). Transcriptome sequencing has the advantage of allowing an experimental analysis of 5′ and 3′ ends of transcripts, and the identification of transcripts from genes that were not predicted to exist. For example, analysis of the transcriptome of Helicobacter pylori using a differential RNA-seq approach revealed considerable antisense transcription, hundreds of candidate small RNA genes and several genes for which the 5′ end was misannotated (Sharma et al., 2010). Similarly, proteomic technologies can be used to verify that predicted proteins are actually made, and identify novel proteins from nonannotated genes (e.g. Kim et al., 2009). Although these approaches do not provide a means by which the existence of a gene can be disproven, their use in refining genome annotations is likely to increase with the falling costs associated with these techniques.
Finally, we strongly suggest that there should be greater consideration of nonconventional analytical approaches to build our understanding of genomes and to solve many of the unanswered functional genomic questions: the recent spate of interest in sRNAs (in Pseudomonas, e.g. Kay et al., 2005; Reimmann et al., 2005; Sonnleitner et al., 2009; Humair et al., 2010) is one marker of evidence that this is progressing, but other avenues need to be explored further. While computational annotations are powerful and able to accurately predict a large number of genes, it is clear that there are some that escape detection. Using modifications of the RNA-seq, which generate a strand-specific output, antisense coding regions can be readily detected in bacterial genomes, greatly enhancing the known pool of antisense genes revealed by genetic means (Silby & Levy, 2004, 2008; Silby et al., 2004; Filiatrault et al., 2010; Sharma et al., 2010). A major frontier in the understanding of the biology of Pseudomonas spp. is the exploration of life outside of the laboratory, and even in nontraditional niches (Stavrinides et al., 2009). A functional genomics approach using emerging sequencing and proteome technology alongside established methods, coupled with the ongoing generation of new genome sequences, will further our understanding of the genus Pseudomonas, and will ensure that we continue to be surprised by these versatile bacteria.
Acknowledgements
We thank the three anonymous reviewers for their excellent suggestions and detailed feedback. M.W.S. and S.B.L. gratefully acknowledge support from the National Research Initiative Competitive Grant no. 2006-35604-16673 and the Agriculture and Food Research Initiative Competitive Grant 2010-65110-20392 from the USDA's National Institute of Food and Agriculture, Microbial Functional Genomics Program. C.W. acknowledges the support of the United Kingdom National Institute for Health Research.
References
Editor:Miguel Camara

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}